
ABSTRACT
Whole-genome sequencing was conducted on two Aspergillus oryzae strains used for the manufacturing of food enzymes, Acrylaway® and Shearzyme®, with the aim of identifying the inserted locus of randomly integrated expression plasmid and obtaining flanking sequences for safety assessment. Illumina paired-end sequencing was employed, and the obtained reads were mapped to two references: the public genome sequence of Aspergillus oryzae RIB40 and the in-house sequence of the used expression plasmid. Introducing the concept of linking-reads, one locus for each was successfully identified as the integrated site. In the case of Acrylaway®, the obtained sequences suggested that the expression plasmid had been integrated as multiple copies in tandem form. In the case of Shearzyme®, however, information on one edge of the insert was missing, which required extra polymerase chain reaction (PCR) cloning for safety assessment. A 4-kb deletion was detected at the integrated site. There was also evidence of rearrangement occurring in Shearzyme® strain.
Graphical Abstract
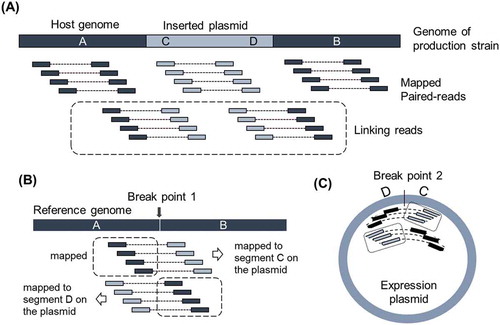
Concept of linking reads made the identification of inserted locus possible.
Enzymes used for food processing are usually considered food additives and/or processing aids. A safety assessment of such enzymes is required in many countries prior to their introduction onto the market [Citation1,Citation2]. Many food enzymes are now manufactured using genetically modified microorganisms [Citation1,Citation3,Citation4], so the safety assessment of new products has drawn increasing attention. The standards of safety assessment of food enzymes differ from one country to another [Citation1–Citation3], but it is usually required to assess the production strains are neither pathogenic nor toxigenic [Citation3,Citation4]; in addition, the details of any genetic modification of the production organism are required for safety assessment when gene technology is applied [Citation1,Citation3–Citation5]. Toxicological studies on the enzyme product are also employed in many countries to confirm its safety [Citation1,Citation4,Citation5]. On the other hand, in some countries, such as Japan, information on the DNA sequence resulting from the modification of a chromosome in production organisms, such as integrated foreign DNA as well as its flanking sequences, is often required to assess the risk of generating unintended harmful compounds based on the sequences. It was previously laborious and costly to obtain such sequence information, but this has dramatically improved due to the development of new technologies such as next-generation sequencing (NGS).
The manufacturing of industrial enzymes using genetically modified microorganisms started in the late 1980s [Citation6–Citation8]. Previously, classical mutagenesis by UV irradiation and/or gamma-ray radiation or by chemical treatment using for example ethyl methane sulfonate or N-methyl-N′-nitro-N-nitrosoguanidine was the standard method of breeding microorganisms [Citation9,Citation10] to improve enzyme productivity. However, using this approach, it took years to develop economical production strains [Citation9]. Fortunately, the application of gene technology has significantly reduced the time and cost required to develop production strains. It has also expanded the range of industrial enzymes available [Citation3,Citation4]. For instance, industrial enzymes used to be exclusively derived from microbes with a strong ability to produce enzymes, such as Bacillus and Aspergillus [Citation11]; however, recombinant technology opened the door to microbes that are usually not easy to grow and even to protein-engineered variants [Citation3,Citation4]. Gene technology has thus become indispensable for the development of new beneficial industrial enzymes.
Certain microorganisms are preferably used for the manufacturing of industrial enzymes, including Bacillus species such as B. subtilis, B. licheniformis, and B. amyloliquefaciens, and fungal species such as Aspergillus oryzae, Aspergillus niger, and Trichoderma reesei [Citation3]. They are all capable of producing enzymes at high levels and have a long history of safe use for food processing and/or the manufacturing of food additives including enzymes. Since the late 1980s, Aspergillus oryzae has been actively used to produce various industrial enzymes [Citation6]. As the standard process, the expression plasmid carrying the gene encoding the particular enzyme was introduced into the host strain by transformation in a circular or linear form [Citation12,Citation13]. In the absence of autonomously replicating sequences, the introduced expression plasmid was integrated into the host genome at random without controlling the integration locus or the gene copy number to be integrated. The resulting transformants varied concerning their enzyme productivity and the copy number of introduced genes [Citation6], and there was no correlation between the gene copy number and enzyme productivity [Citation7]. Moreover, there has been no concrete evidence that a certain locus provides higher enzyme productivity. In conclusion, the best way to isolate the highest enzyme-producing transformant was to generate a large number of transformants by random integration, followed by large-scale screening for their production ability.
In this study, we attempted to identify the integrated locus of randomly inserted expression plasmid in A. oryzae by whole-genome sequencing of two production strains of food enzymes. The challenge in doing so was that the introduced expression plasmid could have been integrated at multiple loci or as multiple copies per locus, which would generate repeated sequences on the genome of production strains and make genome sequencing by NGS difficult [Citation14]. Here, we report our attempt at this in two cases, including estimation of the copy number of the introduced plasmids.
Materials and methods
Strain and DNA preparation
The strains used in this study were A. oryzae pCaHj621/BECh2#10, which is the strain used for the industrial production of asparaginase and originated from Aspergillus oryzae, called Acrylaway® (Novozymes A/S), and A. oryzae pMT2155/BECh2-Fb-3, the strain for producing xylanase from Aspergillus aculeatus, called Shearzyme® (Novozymes A/S). In brief, these strains were generated from a production host strain, A. oryzae BECh2 [Citation15], by transformation with the circular expression plasmid pCaHj621 or pMT2155. These plasmids carry the expression cassette for the enzyme, asparaginase or xylanase, consisting of the coding region of aimed enzyme gene flanked by the neutral amylase promoter and glucoamylase terminator from A. niger, as well as a transformation marker gene, amdS from A. nidulans [Citation16]. Fb-3 is a classical mutant derived from pMT2155/BECh2 by UV irradiation. Details of the construction of pMT2155 can be found in patent application US 9,416,384 B2. The strains were grown in liquid YPD medium (0.5% yeast extract, 1% Bacto peptone, and 2% glucose) at 30ºC for 1 day. Mycelia were harvested by filtration and kept frozen until use. Genomic DNA was extracted from ground frozen mycelia by Automill (Tokken Inc., Japan) and purified using Fast DNA SPIN Kit for Soil (MP Biomedicals) following the manufacturer’s protocol. Over 50 µg of DNA precipitated by ethanol was submitted to Genaris, Inc. (Yokohama, Japan), for whole-genome sequencing.
Genome sequencing
Illumina paired-end library construction and whole-genome sequencing were conducted by Beckman Coulter Genomics (Massachusetts, USA), following the manufacturer’s instructions. Each set of genomic DNA was physically fragmented and the libraries were constructed using fragments of 400–500 bp in length, followed by sequencing by 100 bp from each end. In this study, reads for A. oryzae pCaHj621/BECh2#10 and A. oryzae pMT2155/BECh2-Fb3 are referred to as NZ-A and NZ-S, respectively.
Mapping and analysis
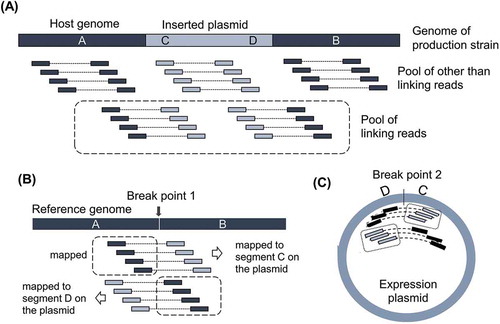
To improve the accuracy of downstream data analysis, reads were filtered and trimmed using the qtrim program (unpublished method) developed by Genaris, Inc. (Yokohama, Japan), in such a manner that maximizes the differences in average quality between trimmed sequences and sequences being trimmed. Obtained reads were mapped onto the reference genome sequence of A. oryzae RIB40 available from DOGAN by NITE (http://www.bio.nite.go.jp/dogan/project/view/AO), as well as onto the sequence of the used expression plasmids, pCaHj621 or pMT2155. Mapping was performed with BWA (Burrows–Wheeler Alignment Tool) [Citation17]. The paired reads containing one read from the A. oryzae genome sequence and another from the plasmid sequence were pooled as linking reads and mapped separately from the pool containing other reads ()). Mapped reads were stored and visualized in GiNeS, a cloud-based platform for analyzing NGS data developed by Genaris, Inc. SNVs and indels detected against the reference sequence were also available in GiNeS.
Figure 1. Idea of identification of breakpoints with linking reads.
(A) Illustration of Pool of other than linking reads to the genome of production strain. Pool of linking reads is defined as the paired read of which one read is from host genome and the other read is from inserted expression plasmid. Thick bar illustrates the genome of production strain around the integration site. A and B are flanking regions of inserted plasmid originated from the host genome. C and D indicate the regions around the edges of inserted expression plasmid. (B) Idea of identification of breakpoint on the host genome by mapping Pool of linking reads to the reference genome. (C) Idea of identification of breakpoint on expression plasmid. Illustration was made assuming the inserted plasmid as a single copy.
Southern blotting analysis
Approximately 3 µg of genomic DNA was digested by a restricted endonuclease for 16 h, then separated on a 0.8% agarose gel. After electrophoresis, the separated DNA in the gel was fragmented with 0.2 M HCl for 15–20 min, denatured in 0.5 M NaOH/1.5 M NaCl for 20–30 min, and neutralized in 1 M Tris/HCl pH 7.5 with 1.5 M NaCl for 15–20 min, followed by transfer to a Hybond N+ (BIO-RAD) in 20× SSC buffer overnight. The DNA was UV-crosslinked to the membrane for 4 min. The membrane was prehybridized for 1 h at 42°C in an appropriate volume of Easyhyb (Roche). The hybridization solution was replaced with a fresh solution containing the denatured DIG-labeled probe and incubated at 42°C, overnight. The membrane was washed twice for 5 min in 2× SSC, 0.1% SDS, at room temperature and twice for 15 min in 0.1× SSC, 0.1% SDS, at 68ºC. Detection of the hybridized bands was performed using DIG DNA detection kit (Roche). The used DNA marker was DIG-labeled DNA molecular weight marker II (Roche). To estimate the copy number, digested DNA was diluted with TE buffer and then applied to an agarose gel. The intensities of the hybridized bands were quantified using Gel DocTM EZ Imager (Bio-Rad).
Sequence confirmation by PCR
The confirmation of sequences around the breakpoints was performed by PCR cloning followed by sequencing. The primers used for the Acrylaway strain were as follows: sp1: TCCGAAATACAGTCATCATAACAC, sp2: TGATACATCGCATCG ACAAGGGAC, sp3: CGCCACCACGAATCCCAACGATCG, and sp4: TTGAGCG GCACTTTCTCTGAACGT, and those used for the Shearzyme strain were: fb1: GAG GCTGCTTCCGCCGACGGAGTG and fb2: GTAGAACCGAAGAGATATGACA CG. The primer fb3: GAGCTCTCCTAGGTATCGGTCCAG was used for genome walking on the Shearzyme strain together with the commercially available adaptors (Takara, Japan) to clone the missing edge of the expression plasmid. The amplified DNA fragments were purified and TA-cloned with the TOPO DNA cloning kit (Invitrogen). The sequencing of the amplified DNA was outsourced to Hokkaido System Science, Inc. (Sapporo, Japan).
Results and discussion
Sequencing, trimming and analysis of reads
Sequencing and mapping data are summarized in . In total, over 7500 Mb of sequences were read for each strain, which was equivalent to 200× coverage of the whole genome size of A. oryzae (ca. 38 Mb). Cleaning of reads was performed to improve the accuracy before mapping. Figure S1 shows the average and standard deviation of the quality at each base position of the reads before and after the filtering and trimming processing. The average quality improved and became consistent at each base position of the reads. Furthermore, the standard deviation decreased significantly after trimming, indicating that the read filtering and trimming had worked well.
Table 1. Analysis of reads from Illumina paired-end library.
After cleaning, over 99% of reads were successfully mapped to either reference and over 97% of reads were properly paired. Mapped reads were grouped into two pools: one was the pool of linking reads and the other was the pool of other reads ()). The frequency of linking reads was around 0.02% of total reads in both cases. There were over 3000 variations relative to the reference genome, such as SNVs and indels, on each of NZ-A and NZ-S relative to the reference genome. These variants must have arisen from the difference between the host strain BECh2 and the reference strain RIB40 or been generated during the transformation event and/or post-transformation modification, if any.
Mapping of linking reads
Upon evaluating the mapping results of linking reads on GiNeS, there were three common regions where the linking reads from both NZ-A and NZ-S were clustered onto the reference genome sequence (, Figure S2–S4). The expression plasmids were constructed using the DNA fragments isolated from Aspergillus, therefore, it was predicted that the mapped reads were originated from the used expression plasmids which could be homologues to the sequence on A. oryzae genome. As expected, two regions were 5′-UTR regions of alpha-amylase genes, AO090120000196 and AO090023000944, which is highly homologous to the promoter used in the expression plasmids, derived from the neutral amylase gene of Aspergillus niger. Another region was 1.9 kb downstream of the presumed maltase/glucoamylase gene AO090003001209 (Figure S4); however, it was difficult to explain how the mapped region was related to the DNA used for the expression plasmids. Upon closer examination of the sequence of the reference, it was surprisingly found that the mapped region was also highly homologous to the promoter used in the expression plasmid. When investigating the sequence further, one more alpha-amylase gene, which had not been annotated at DOGAN, was discovered downstream of this region. The sequence alignment with two other alpha-amylases is shown in Figure S5. Whether the discovered gene is functional and encodes the active alpha-amylase needs to be investigated, but at least it was confirmed that A. oryzae RIB40 has three copies of homologous alpha-amylase genes, as was previously reported [Citation18]. These three commonly mapped regions were carefully investigated about the possibility of presence of breakpoints, but no evidence of integration of expression plasmids was found.
Table 2. Loci on the reference genome where linking reads were mapped to.
Besides these common regions, there were mapped clusters specific to either NZ-A or NZ-S. In the case of NZ-A, one of the such regions was AO090005000816 encoding asparaginase (Figure S6). Notably, the reads were mapped only to exons but not to introns, as the expression plasmid carried the cDNA clone of the asparaginase gene from A. oryzae after its introns spliced. In the case of NZ-S, there was one region to which only 200 bp of linking reads were mapped (Figure S7). This could be an artifact related to the used xylanase gene, but it was unclear. These regions were carefully examined for the possibility of containing breakpoints, but no evidence of integration of the expression plasmids in these regions was identified. The remaining mapped regions were further investigated to determine the possibility of integration.
Identification of potential inserted locus
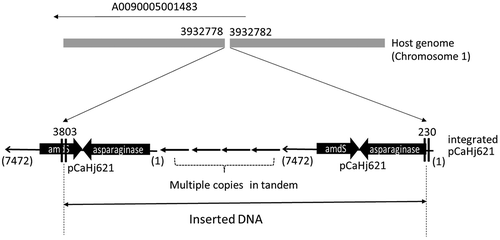
NZ-A: In the case of NZ-A, there was a cluster of mapped reads onto SC005 on chromosome 1 around the 5′-end of coding region AO090005001483 (). The gene was not related to the sequences used in the expression plasmid. Upon close examination of the mapped reads, a unique point was discovered around position 3932800, with the directions of mapped reads being opposite before and after this point ()). A similar result was seen for the reads without linking reads at the same position ()). Moreover, the sequences on these reads that were not mapped to the reference genome, shown in gray in ), corresponded to the sequence of the used expression plasmid pCaHj621. From these results, it was concluded that the expression plasmid was integrated at the position between 3932778 and 3932782 on reference chromosome 1. One of the edges of the insert was at position 230 of pCaHj621, which was in the promoter region of expression cassette, and the other was located at position 3803 in the middle of the marker gene amdS. Both regions need to be functional for successful transformation event and production of the asparaginase. Therefore, the result indicated that the expression plasmid had been integrated as multiple copies in tandem form, so that at least one functional set of expression plasmid exist in the insert. Further details of the insert structure could not be elucidated from the genome sequencing.
Figure 2. View on GiNeS around the inserted locus of the expression plasmid pCaHj621.
Mapping results of four different pools, 1)~4), toward the reference A. oryzae RIB40 are shown. Pink bars represent depth of coverage of reads at each base position. Genes show the annotated CDS in DOGAN and arrows in parenthesis indicate the direction of CDS. What is shown in GiNeS is explained in more detail in the supplemental Figure S2. A cluster of linking reads unique to NZ-A is indicated by a circle in broken line. Predicted integration locus of the expression plasmid pCaHj621 is shown with red arrows. It was close to the 5ʹ end of gene AO09005001483 on chromosome 1. Linking reads from NZ-S were not mapped to this locus.
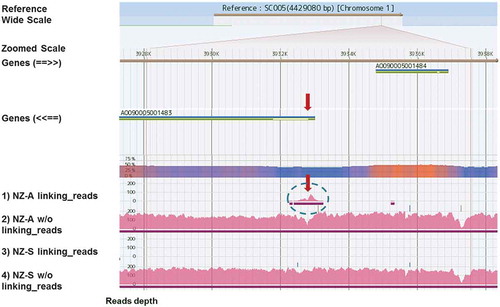
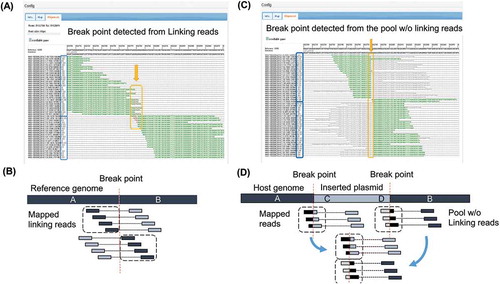
Figure 3. Detected breakpoints and mapped reads.
(A) View of the mapped linking reads from NZ-A around the predicted integration locus. Identified breakpoint is shown with arrow. Sequences shown in green match to the reference genome sequence, while mismatched bases are shown in red. Direction of mapped reads, indicated by (+) or (-) in blue boxes, become opposite on the upper half and the lower half of mapped reads. (B) Illustration of mapped linking reads shown in (A). (C) View of the mapped reads from the pool of NZ-A without linking reads. Identified breakpoint is shown with arrow. Sequences shown in green match to the reference genome sequence. Sequences of soft-clipped portion of mapped reads correspond to the sequence of the expression plasmid. Direction of the mapped reads become opposite on the upper half and the lower half of mapped reads. (D) Illustration of mapped reads shown in (C). Magnified figures of (A) and (C) are available in Supplemental Data.
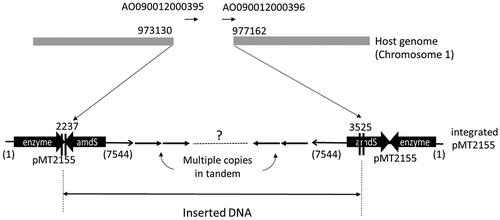
NZ-S: The potential inserted locus for NZ-S found on GiNeS is shown in Figure S8. It was on reference chromosome 4 and accompanied by a deletion of a 4-kb-long region. The deleted region contained the whole of the AO090012000395 gene annotated as a soluble epoxide hydrolase and the N-terminal part of AO090012000396, the function of which is unknown. In a similar manner to the previous case, one of the edges of the insert located next to the genome reference position 973130 was successfully identified from the reads. It was at position 2237 of pMT2155 and within the terminator of expression cassette. On the other hand, sequence information of the other edge of the insert connected to the genome at reference position 977162 was missing in the reads. Without the information on the edge of the insert, assessment based on the sequences was impossible. It was considered that an alternative assessment was necessary in this case.
Southern blotting analysis
Southern blotting analysis was conducted to estimate the structure of the insert, namely, how the introduced plasmid was integrated into the chromosome. The genomic DNA of the Acrylaway® production strain A. oryzae pCaHj621/BECh2#10 was digested with BamHI alone or together with NsiI and hybridized with the coding region of the asparaginase gene. BamHI digestion solely generated a band of an endogenous asparaginase gene of 5.6 kb, as seen on the host strain BECh2 (lane 10, Figure S9(B)). On the other hand, the production strain had two extra bands of 7.5 and 3.5 kb, with stronger intensity on the band of 7.5 kb (lane 11, Figure S9(B)). The size of the expression plasmid pCaHj621 was 7.5 kb. As the plasmid pCaHj621 had a single BamHI site, the strong signal of 7.5 kb indicated that the plasmid was integrated as multiple copies in tandem form, as described in the literature [Citation12,Citation13]. The 3.5 kb band must be the fragment containing a border site. As no other bands were observed, it was concluded that the plasmid was integrated at a single site as multiple copies in tandem form with no rearrangement.
Integrated copy numbers were estimated by dilution of DNA on Southern blotting using the samples subjected to double digestion with BamHI and NsiI. The host strain BECh2 carried asparaginase as a single copy; therefore, the band of 2.3 kb from BECh2 (lane 2, Figure S9(C)) was used as a reference. The integrated expression plasmid pCaHj621 generated a band of 1.5 kb. By comparing the intensities of this 1.5 kb band and the reference, it was estimated that the production strain carried around eight copies of the expression plasmid. This was also supported by a comparison of the intensity of the bands from sole BamHI digestion (5.6 kb band in lane 10 and 7.5 kb band in lane 11, Figure S9(C)).
Southern blot analysis was also performed on the production strain of Shearzyme®, A. oryzae pMT2155/BECh2-Fb3 (Figure S10). The donor strain of the xylanase gene, A. aculeatus NV-132, was used as a reference. Genomic DNA of the strains was digested with EcoRI, HindIII, and XhoI together and hybridized with the xylanase gene as a probe. The reference showed a 1.8 kb band of the xylanase gene corresponding to a single copy of this gene. Compared with the intensity with diluted DNA of the production strain, it was estimated that the production strain carried approximately 30 copies of the expression plasmid. The DNAs of the production strain and the host strain were digested with EcoRI, cleavage site for which exists only one at pMT2155, and hybridized with the xylanase gene. The host strain BECh2 did not produce any signal, but the production strain showed a 7.5 kb band with the strongest intensity. This corresponded to the size of the expression plasmid pMT2155; this indicated that the plasmid was integrated as multiple copies in tandem form. There were a couple of extra bands for the production strain; however, it was not possible to elucidate more about the structure of the insert in this experiment. All in all, the results indicated that the expression plasmid was integrated about 30 copies in tandem form.
Confirmation by PCR
Integration of pCaHj621 in A. oryzae pCaHj621/BECh2#10 at the identified locus was confirmed by PCR cloning followed by sequencing. The fragments including a border on each edge were amplified using the primer sets sp1/sp2 and sp3/sp4 and subjected to sequencing. The obtained sequences perfectly matched with the prediction, as determined by genome sequencing. It was confirmed that the plasmid pCaHj621 had been integrated into the locus AO0090005001483 with the deletion of 3-bp, as shown in . One of the edges of the insert was in the promoter region of the expression plasmid and the other edge was within amdS. The elucidated structure of the insert is illustrated in . Southern blotting analysis suggested that eight copies in total of the expression plasmid had been integrated in tandem form. It was a good agreement with the observation previously reported by others [Citation12,Citation13] that in the case of fungal transformation event, introduced plasmid was integrated as multi-copies in tandem array at ectopic loci.
Figure 4. Identified inserted locus of pCaHj621 and structure of integrated plasmid.
The breakpoints at host genome and the edges of insert are shown with the position numbers from the references. Deletion of three bases was observed at the inserted locus on the host genome. One of the edges of integrated plasmid at 230 was in the promoter region of the expression cassette for asparaginase. Another edge at 3803 was in the middle of marker gene, amdS. As at least one functional set of expression plasmid is necessary, it was predicted that the expression plasmid pCaHj621 was integrated as multiple copies in tandem form. Southern blotting analysis revealed that the strain carried 8 copies of pCaHj621.
The border sequence of the identified edge of pMT2155 in A. oryzae pMT2155/BECh2-Fb3 was also confirmed by PCR. The result showed that the edge at reference position 973130 on the chromosome was connected to position 2237 of the expression plasmid pMT2155, as predicted (). Another edge of the insert was missing in the genome sequencing; therefore, cloning by genome walking was attempted to identify the missing edge. A fragment of about 2.3 kb containing the border was successfully isolated, and its sequence information revealed that another edge of the insert was at position 3525, close to the 3′ end of amdS on pMT2155 (). Interestingly, the sequence direction of the integrated expression plasmid at this edge was opposite to that at the other edge. Considering the introduced plasmid is usually integrated in tandem manner, the result indicated that rearrangement had occurred in the inserted region after integration of plasmid and made the direction of the integrated plasmid opposite in some part of the insert. From Southern blotting analysis, it was estimated that the strain carried approximately 30 copies of the expression plasmid in tandem form. The high copy numbers in tandem at one locus might have high chance of rearrangement within the insert. Furthermore, the classical mutagenesis event to generate Fb-3 could be another cause of the rearrangement in the insert, as well as the cause of 4-kb deletion at the integration site. In any case, further elucidation of the insert structure was not possible in this experiment.
Figure 5. Identified inserted locus of pMT2155 and illustration of integrated plasmid.
The breakpoints at the host genome were shown with the position number of reference genome. 4-kb long fragment was deleted between breakpoints. One of the edges of inserted plasmid pMT2155, at 2237, was determined by genome sequencing. It was within the terminator region of expression cassette of xylanase gene. Another edge, at 3525, was identified by PCR cloning. It was around 5ʹ-end of amdS. Southern blotting analysis indicated around 30 copies of plasmid was integrated in tandem form. However, directions of the integrated plasmid at both edges were opposite, that indicated the rearrangement occurred in the insert. Further structure of the insert is not known.
Conclusion
The locus of the randomly integrated expression plasmid inserted into the chromosome of A. oryzae strain was successfully identified by whole-genome sequencing with Illumina paired-ends in two cases. The challenge was that the potential repeated sequences caused by multi-copy integration and/or multi-locus integration of the introduced expression plasmid could disturb the assembly of short reads and make identification of the integration locus difficult. Fortunately, in both cases, the event of integration of the expression plasmid occurred at one locus on the chromosome. They were non-homologous integration at loci non- relating to the introduced plasmid sequences. With the concept of linking reads, it was possible to identify the integration locus precisely as breakpoints on the host chromosome. Integrated locus for asparaginase was near 5ʹ end of AO090005001483 presumed glycosyl transferase gene on chromosome 1 and the integration site for xylanase was between 973130 and 977162 on chromosome 4 by the position numbers of reference genome. There results provided information of flanking sequences of the inserts on the production strains, which is often required for safety assessment.
Genome sequencing successfully identified the breakpoints on the expression plasmid in case of Acrylaway®, which made the sequence-based safety assessment possible on its production strain by combining flanking sequences information. Including the results from Southern blotting analysis, the structure of the insert was estimated as shown in . Integrated copy numbers of expression plasmid were estimated as 8 copies. On the other hand, in the case of Shearzyme®, additional cloning work other than genome sequencing was necessary to identify the breakpoints on the expression plasmid. This might be related to the rearrangement occurred on the chromosome around the integration locus, accompanied with a 4-kb deletion on the host chromosome. Moreover, there was another indication of rearrangement within the insert, i.e. the direction of integrated expression plasmid was opposite at two edges of the insert. Whether this rearrangement occurred during or after the event of integration of the expression plasmid or was caused by classical mutagenesis on the primary transformant was not known. The estimated structure of the insert is shown in . Integrated copy number was estimated about 30 copies. Further elucidation of the structure of the insert was not possible in this work.
This time only two cases were studied to predict the integration locus of introduced plasmid as well as the structure of insert, but in both cases, the tested strains got integration at one locus with non-homologous integration event with multi-copies integration in tandem form. It was a good agreement with previously reported observation as fungal transformation [Citation12,Citation13]. The tested strains were selected as high-per producing strains of these enzyme, but interestingly, the integrated copy numbers were varied; one was 8 copies and the other was 30 copies. Integration loci were totally different between two cases. In that sense, it can be said that the knowledge about the strain structure to get high-per enzyme productivity is still limited.
Author contribution
A.T. established the locus identification method and summarized the data at Genaris, H.U. made follow-up studies, S.T. initiated the project, performed the analysis of data from Genaris, and prepared the manuscript, Y.U. supervised the project and reviewed the manuscript.
Supplemental_Data_-revision_draft.docx
Download MS Word (1.4 MB)Acknowledgments
We thank for Mayuko Kodama for technical support, Mikako Sasa and Enago (www.enago.jp) for language editing, Carsten Hjort for the advice and approval of publication.
Disclosure statement
S.T. and H.U. are present employee of Novozymes Japan Ltd. A.T. and Y.U. are former employee of Genraris Inc. Locus identification by genome sequencing was out-sourced from Novozymes to Genaris.
Supplementary material
Supplementary data for this article can be accessed here.
Related Research Data
References
- Spök A. Safety regulations of food enzymes. Food Technol Biotechnol. 2006;44:197–209.
- Magnuson B, Munro I, Abbot P, et al Review of the regulation and safety assessment of food substances in various countries and jurisdictions. Food Addit Contam Part A. 2013;30:1147–1220.
- Olempska-Beer ZS, Merker RI, Ditto MD, et al. Food-processing enzymes from recombinant microorganisms – a review. Regul Toxicol Pharmacol. 2006;45:144–158.
- Pariza MW, Johnson EA. Evaluating the safety of microbial enzyme preparation used in food processing: update for a new century. Regul Toxicol Pharmacol. 2001;33:173–186.
- EFSA. Panel on Genetically Modified Organisms (GMO). Scientific opinion on guidance on the risk assessment of genetically modified microorganisms and their products intended for food and feed use. EFSA J. 2011;9:2193.
- Christensen T. Application: Aspergillus oryzae as a host for production of industrial enzymes. In: Powell KA, Renwick A, Peberdy JF, editors. The genus aspergillus. From taxonomy and genetics to industrial application. New York (NY): Plenum Press; 1994. p. 251–259. (Federation of European Microbiological Societies Symposium Series: Volume 69).
- Christensen T, Woeldike H, Boel E, et al High level expression of recombinant genes in Aspergillus oryzae. Bio/Technology. 1988;6:1419–1422.
- Sasa M, Takagi S. The application of Aspergillus oryzae in industry. In: Watanabe MM, Suzuki K, Seki T, editors. Innovative roles of biological resource centers. Proceedings of the tenth international congress for culture collections. Tsukuba, Japan: Japan Society of Culture Collections & World Federation for Culture Collections; 2004 Oct 10-15. p. 29–35.
- Calam CT. Improvement of micro-organisms by mutation, Hybridization and selection. In: Norris JR, Ribbons DW, editors. Methods in microbiology. Vols. 3, Part A. New York (NY): Academic Press; 1970. p. 435–459.
- Volesky B, Luong JHT, Aunstrup K. Microbial enzymes: production, purification, and isolation. Crit Rev Biotechnol. 1984;2:119–146.
- Aunstrup K. Production, isolation, and economics of extracellular enzymes. In: Wingard L, editor. Applied biochemistry and bioengineering. enzyme technology. Vol. 2. New York (NY): Academic Press; 1979. p. 27–69.
- Fincham JRS. Transformation in Fungi. Microbiol Rev. 1989;53:148–170.
- Yelton MM, Hamer JE, Timberlake WE. Transformation of Aspergillus nidulans by using a trpC plasmid. Proc Natl Acad Sci USA. 1984;81:1470–1474.
- Schatz MC, Delcher AL, Salzberg SL. Assembly of large genomes using second-generation sequencing. Genome Res. 2010;20:1165–1173.
- Christensen BE, Inventor; Novozymes A/S, assignee. Methods for producing polypeptides in Aspergillus mutant cells. PCT application WO00/39322. 2000 Jul 6.
- Hynes MJ, Corrick CM, King JA. Isolation of genomic clones containing amdS gene of Aspergillus nidulans and their use in the analysis of structural and regulatory mutations. Mol Cell Biol. 1983;3:1430–1439.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760.
- Machida M, Asai K, Sano M, et al Genome sequencing and analysis of Aspergillus oryzae. Nature. 2005;438:1157–1161.