
ABSTRACT
The orientation of the three domains in the bifunctional aspartate kinase-homoserine dehydrogenase (AK-HseDH) homologue found in Thermotoga maritima totally differs from those observed in previously known AK-HseDHs; the domains line up in the order HseDH, AK, and regulatory domain. In the present study, the enzyme produced in Escherichia coli was characterized. The enzyme exhibited substantial activities of both AK and HseDH. L-Threonine inhibits AK activity in a cooperative manner, similar to that of Arabidopsis thaliana AK-HseDH. However, the concentration required to inhibit the activity was much lower (K0.5 = 37 μM) than that needed to inhibit the A. thaliana enzyme (K0.5 = 500 μM). In contrast to A. thaliana AK-HseDH, Hse oxidation of the T. maritima enzyme was almost impervious to inhibition by L-threonine. Amino acid sequence comparison indicates that the distinctive sequence of the regulatory domain in T. maritima AK-HseDH is likely responsible for the unique sensitivity to L-threonine.
Abbreviations: AK: aspartate kinase; HseDH: homoserine dehydrogenase; AK–HseDH: bifunctional aspartate kinase–homoserine dehydrogenase; AsaDH: aspartate–β–semialdehyde dehydrogenase; ACT: aspartate kinases (A), chorismate mutases (C), and prephenate dehydrogenases (TyrA, T).
Graphical Abstract
The bifunctional aspartate kinase-homoserine dehydrogenase (AK-HseDH) from Thermotoga maritima is a novel type of the L-threonine-sensitive AK-HseDH.
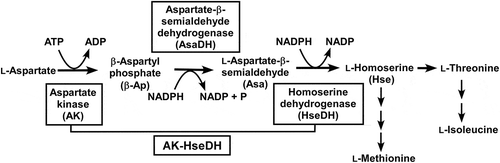
Aspartate kinase (AK, EC 2.7.2.4) and homoserine dehydrogenase (HseDH, EC 1.1.1.3) are involved in the biosynthetic pathway from L-aspartate to L-homoserine (Hse) in plants and microorganisms (). Hse is a common precursor for the synthesis of L-methionine, L-threonine, and L-isoleucine. At the first step in this pathway, L-aspartate is phosphorylated to β-aspartyl phosphate (β-Ap) by AK. The second enzyme, aspartate-β-semialdehyde dehydrogenase (AsaDH, EC 1.2.1.11), converts β-Ap to L-aspartate-β-semialdehyde (Asa). The third enzyme, HseDH, catalyzes the NADPH-dependent reduction of Asa to Hse. Subsequent metabolisms of Hse yield L-methionine, L-threonine, and L-isoleucine, which are all essential in humans. These pathways have been known to be highly regulated by the end products, and in particular, L-threonine-dependent feedback inhibition to control the activities of AK and HseDH has been analyzed in detail [Citation1–Citation5]. Both enzymes are important as potential targets for the structure-based designing of antibiotics or herbicides because the enzymes are not present in mammals [Citation6–Citation8], as well as for improving the nutritional value of food crops [Citation9–Citation12].
Figure 1. Biosynthetic pathway from L-aspartate to L-homoserine.
In bacteria and plants, AK and HseDH function as either a monofunctional enzyme or fused-bifunctional enzyme (AK-HseDH). In Escherichia coli and Arabidopsis thaliana, L-threonine-sensitive AK-HseDHs are present [Citation13–Citation15]. A linker domain between the AK catalytic and the HseDH catalytic domains has been reported to be responsible for the regulatory effect of L-threonine () [Citation15–Citation19]. This intermediate regulatory domain is composed of two homologous subdomains [Citation17]; the two ACT subdomains that are the structural feature of a number of allosteric enzymes including AKs (A), chorismate mutases (C), and prephenate dehydrogenases (TyrA, T). [Citation20] The ACT subdomain includes secondary structures of the loop-α helix-loop-β strand-loop-β strand motif (loop-α-loop-β-loop-β motif) and can be found in one copy or in duplicate (in the case of AK-HseDH). Based on the structure prediction of the two ACT subdomains of A. thaliana AK-HseDH, two potential L-threonine-binding sites have been identified, and the mechanism of the L-threonine-dependent inhibition has been proposed [Citation13]. However, information on the inhibition mechanism remains limited because the structure of bifunctional AK-HseDH has not yet been determined.
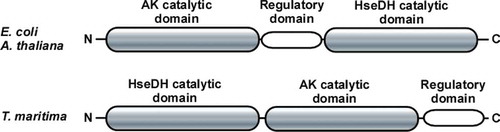
Figure 2. Orientation of the three domains in AK-HseDHs. Upper panel corresponds to E. coli and A. thaliana AK-HseDHs and lower panel corresponds to T. maritima AK-HseDH.
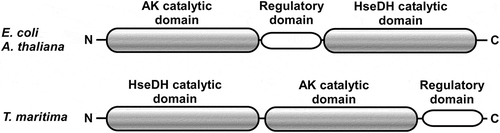
Up to now, no AK-HseDH from hyperthermophiles has been characterized. Hyperthermophilic enzymes are generally much more stable than their counterparts from mesophiles, which permits more detailed examination of their structural features and functionality. Within the genomic sequence of the hyperthermophilic bacterium, Thermotoga maritima, a fused gene encoding both AK and HseDH homologues (open reading frame identification number TM0547) is present [Citation21]. Notably, the predicted orientation of the AK, regulatory, and HseDH domains in the protein encoded by TM0547 totally differs from those observed in E. coli and A. thaliana AK-HseDHs, and the domains line up in the order HseDH, AK, and regulatory domain (). As the first step toward the structural characterization of AK-HseDH, we expressed that gene in E. coli, characterized the enzyme produced, and revealed the enzyme to be a novel type of the L-threonine-sensitive AK-HseDH. This study is the first to describe the presence of AK-HseDH in hyperthermophiles.
Materials and methods
Expression and purification of AK-HseDH
The gene encoding the T. maritima AK-HseDH homologue (TM0547, the gene information is available in the Kyoto Encyclopedia of Genes and Genomes database) was amplified using PCR. The oligonucleotide primers used to amplify the gene fragment were 5’‑TTAGGATCATATGAGAACATATCGATCC-3’, which contains a unique NdeI restriction site (bold) overlapping the 5’ initiation codon, and 5’‑CCCAGTCGACTCACTCCCTGTCCAGTT-3’, which contains a unique SalI restriction site (bold) proximal to the 3’ end of the termination codon (underlined).
Chromosomal T. maritima DNA (43589D) was obtained from the American Type Culture Collection (Manassas, VA, USA) and used as the template. The amplified 2.2-kb fragment was digested with NdeI and SalI and ligated with the expression vector pET21b (Novagen, Madison, WI, USA) previously linearized with NdeI and SalI to generate pET21/TM0547, the expression system for a non-tagged enzyme, which was then used to transform the E. coli strain BL21 (DE3) codon plus-RIPL (Agilent Technologies).
The transformants were cultivated at 37°C in 2.0 L of Luria-Bertani medium containing 100 μg/mL ampicillin until the optical density at 600 nm reached 0.7. Expression was induced by adding 1.0 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) to the medium and cultivation was continued for an additional 3 h at 37°C. Cells were then harvested by centrifugation, suspended in 10 mM Hepes buffer (pH 7.5) containing 0.2 M KCl (buffer A), and disrupted by sonication, after which the cell debris was removed by centrifugation (15,000 × g for 30 min).
The resulting supernatant, which served as the crude extract, was heated at 85°C for 10 min, and the denatured proteins were removed by centrifugation (15,000 × g for 30 min). Subsequently, solid (NH4)2SO4 was added to 30% saturation to the supernatant. The resulting enzyme solution was applied onto a Toyopearl Ether-650M column (Tosoh, Tokyo, Japan) previously equilibrated with buffer A supplemented with 30% (NH4)2SO4, and the column was washed with the same buffer. The enzyme was eluted without absorption. The active fractions in the flow through were pooled, concentrated, and loaded onto a Superdex 200 26/60 column (GE Healthcare, Uppsala, Sweden) previously equilibrated with buffer A and eluted with the same buffer. The active fractions were collected and placed on a Cellulofine HAP hydroxyl apatite column (Seikagaku Corporation, Japan) previously equilibrated with 10 mM potassium phosphate buffer containing 0.2 M KCl. The column was washed with the same buffer, and the enzyme was eluted with a linear gradient of 10 to 400 mM potassium phosphate buffer containing 0.2 M KCl. The buffer in the eluted enzyme solution was replaced with buffer A using a combination of dilution and concentration by ultrafiltration (Amicon Ultra 30K NMWL; Millipore, Tokyo). All the purification steps were performed at room temperature.
Expression and purification of AsaDH
The gene encoding the T. maritima AsaDH homologue (TM1523) was amplified by PCR using the same method as described for T. maritima AK-HseDH. The oligonucleotide primers used to amplify the gene fragment were 5’‑ACATATGAAGGTGAAAGTAGGAGTTGTT-3’, which contains a unique NdeI restriction site (bold) overlapping the 5’ initiation codon, and 5’‑AAAGCTTCACACCTTTCCGTTCATC-3’, which contains a unique HindIII restriction site (bold) overlapping the 3’ end of the termination codon (underlined). The amplified 1.0-kb fragment was digested with NdeI and HindIII and ligated with the expression vector pColdIV (Takara Bio, Japan) previously linearized with NdeI and HindIII to generate pColdIV/TM1523, the expression system for a non-tagged enzyme, which was then used to transform the E. coli strain BL21 (DE3) codon plus-RIPL (Agilent Technologies).
The transformants were cultivated at 37°C in 0.5 L of SB medium (1.2% tryptone peptone, 2.4% yeast extract, 1.25% K2HPO4, 0.38% KH2PO4, and 0.5% glycerol) containing 100 μg/mL ampicillin. After 6 h, the culture was cooled to 15°C, IPTG (0.1 mM) was added, and cultivation was continued for an additional 20 h at 15°C. The cells were harvested by centrifugation, suspended in 10 mM Tris-HCl buffer (pH 8.0) (buffer B) and disrupted by sonication, after which the cell debris was removed by centrifugation (15,000 × g for 20 min).
The resulting supernatant was heated at 70°C for 15 min, and the denatured proteins were removed by centrifugation (15,000 × g for 30 min). The supernatant was loaded onto a Toyopearl SuperQ-650M column (Tosoh) that had been equilibrated with buffer B. The column was then washed with the same buffer, and the enzyme was eluted with a linear gradient of 0–0.5 M KCl in the buffer. The AsaDH-containing fractions were checked using SDS-PAGE, pooled, concentrated, and loaded onto a Superdex 200 26/60 column (GE Healthcare) previously equilibrated with buffer B containing 0.2 M KCl and eluted with the same buffer. The buffer in the eluted enzyme solution was changed to 10 mM Hepes buffer (pH 7.5) containing 0.2 M KCl by several cycles of concentration and dilution using an ultrafiltration unit (Amicon Ultra 30K NMWL; Millipore). All the purification steps were performed at room temperature.
Determination of enzyme activity, kinetic parameters, and protein concentration
Enzyme activity was assayed spectrophotometrically using a Shimadzu UV-mini 1240 spectrophotometer (Shimadzu, Kyoto, Japan) equipped with a thermostat. The standard reaction mixture for HseDH (Hse oxidation) consisted of 200 mM Glycine-NaOH buffer (pH 10.5) containing 50 mM Hse, 2.5 mM NADP, 200 mM KCl, and the enzyme in a final volume of 1.0 mL. After warming the reaction mixture by incubation for 3 min at 55°C without the enzyme, the reaction was started by adding the enzyme. The appearance of NADPH was monitored from the absorbance at 340 nm (extinction coefficient ε = 6.22 mM−1⋅cm−1).
AK activity (β-Ap production) was basically determined by measuring the amount of ADP formed from ATP using the coupling reaction with NADH, pyruvate kinase, and lactate dehydrogenase. The standard reaction mixture consisted of 100 mM Hepes-NaOH buffer (pH 7.0), 200 mM KCl, 4 mM magnesium acetate, 45 mM L-aspartate, 4 mM ATP, 1 mM phosphoenolpyruvate, 20 units pyruvate kinase (rabbit muscle), 40 units L-lactate dehydrogenase (rabbit muscle), 0.14 mM NADH, and the enzyme in a final volume of 1.0 mL. After warming the reaction mixture by incubation for 3 min at 55°C without L-aspartate, the reaction was started by adding L-aspartate. The disappearance of NADH was monitored from the absorbance at 340 nm. To determine the kinetic parameters, the initial velocity was examined by varying the concentration of one substrate while keeping the concentrations of the other substrates constant, as previously described [Citation22]. The protein concentration was determined using the Bradford method, with bovine serum albumin serving as the standard [Citation23].
Polyacrylamide gel electrophoresis and molecular mass determination
SDS-PAGE (12.5% acrylamide slab gel, 1 mm thick) was carried out using the procedure of Laemmli [Citation24], after which the protein band was stained with Coomassie Brilliant Blue R-250. The molecular mass of the purified enzyme was determined using a Superdex 200 10/300 GL column (GE Healthcare) with 10 mM Hepes buffer (pH 7.0) containing 0.2 M KCl as the elution buffer. Gel filtration standards (Bio-Rad Lab., Hercules, CA, USA) were used as the molecular mass standards. The subunit molecular mass was determined by SDS-PAGE using nine marker proteins (6.5–200 kDa) (Takara Bio).
Effects of temperature and pH on enzyme stability and activity
The optimal temperature for the HseDH activity was determined by performing the standard assay at temperatures ranging from 50 to 80°C. To determine the effect of temperature on its stability, the enzyme was incubated for 10 min at different temperatures in 10 mM Hepes buffer (pH 7.5) containing 0.2 M KCl. After centrifugation (15,000 × g for 5 min), the residual activity in the supernatant was determined using the standard assay method for HseDH. To determine the effect of pH on its stability, the enzyme was incubated in different pH buffers for 10 min at 50°C, and the remaining activity was again determined under the standard assay conditions for HseDH. The buffers (250 mM) used for these assays were acetate-Na acetate (pH 4.5–6.0), KH2PO4-K2HPO4 (pH 6.0–7.5), glycylglycine-NaOH (pH 7.5–9.0), glycine-NaOH (pH 9.0–10.5), and K2HPO4-K3PO4 (pH 10.5–12.0). To determine the optimal pH for HseDH activity, glycine-NaOH (pH 8.9–11.1) buffer (100 mM) was used in the standard assay method.
To determine the optimal temperature for AK activity, the enzyme activity was assayed by the endpoint method with pyruvate kinase and lactate dehydrogenase. The reaction mixture contained 100 mM Hepes-NaOH buffer (pH 7.5), 200 mM KCl, 4 mM magnesium acetate, 45 mM L-aspartate, 4 mM ATP, and the enzyme in a total volume of 400 μL. The reaction was run at different temperatures for 10 min and then terminated by adding 50 μL of 50% (w/v) trichloroacetic acid, followed by neutralization with 60 μL of 4 M NaOH. The mixture was then centrifuged, and the resulting supernatant was used for the second reaction. The second reaction mixture consisted of 200 mM Tris-HCl buffer (pH 7.5), 1 mM phosphoenolpyruvate, 20 units pyruvate kinase (rabbit muscle), 40 units L-lactate dehydrogenase (rabbit muscle), 0.14 mM NADH, and the supernatant in a final volume of 1.0 mL. After incubation for 10 min at 37°C, ADP concentration was determined from a standard curve based on the change in absorbance at 340 nm. To determine the optimal pH for AK activity, Bis-Tris (pH 6.3–6.8), Hepes (6.5–7.6), and Tris-HCl (pH 7.6–8.4) buffers (100 mM) were used at 55°C in the endpoint method.
Coupled assay of AK-HseDH and AsaDH, and determination of Hse
The overall conversion of L-aspartate to Hse was examined by measuring oxidation of NADPH by AsaDH and HseDH in the presence of T. maritima AsaDH and AK-HseDH. To determine the stoichiometry of the two enzymes, different amounts of T. maritima AsaDH (13.2–198 pmol) were incubated with a fixed amount of T. maritima AK-HseDH (63.4 pmol). The reaction mixture consisted of 100 mM Hepes buffer (pH 8.0), 100 mM KCl, 4 mM magnesium acetate, 15 mM L-aspartate, 2 mM ATP, 0.1 mM NADPH, and the mixture of T. maritima AsaDH and AK-HseDH in a final volume of 1.0 mL. After warming the reaction mixture by incubation for 3 min at 55°C without the enzymes, the reaction was started by adding the enzymes. The disappearance of NADPH was monitored spectrophotometrically from the absorbance at 340 nm.
To determine the amounts of produced Hse, 6.34 pmol of T. maritima AK-HseDH was mixed with 6.60 pmol of T. maritima AsaDH, and the reaction was started as described above. At 5-min intervals, the reaction was terminated by cooling on ice, followed by deproteinization using an ultrafiltration unit (Amicon Ultra 3K NMWL; Millipore). In these assays, the oxidation of NADPH in the reaction mixture was also monitored using a spectrophotometer. The amount of Hse in the filtrate was determined using an ultraperformance liquid-chromatography (UPLC) system (Waters, Milford, MA, USA). Derivatization of Hse was carried out using o-phthalaldehyde (OPA) and N-acetyl-L-cysteine (NAC). A methanolic solution containing 8 mg of OPA and 10 mg of NAC in 1 mL of methanol was prepared and derivatization was performed in the reaction mixture (250 μL) consisting of 25 μL of sample, 50 μL of methanolic solution, and 175 μL of 0.4 M borate-NaOH buffer (pH 10.4). After incubation for 2 min at room temperature in the dark, aliquots (1 μL) of the reaction mixture were introduced into an UPLC system equipped with a Waters AccQ-Tag Ultra 2.1 × 100 mm column (Waters), a Waters Binary Solvent Manager, a Waters Sample Manager, and a Waters FLR Detector. The excitation and emission wavelengths for fluorescent detection were 350 nm and 450 nm, respectively. The system was operated at a flow rate of 0.25 mL/min at 30°C. The UPLC gradient system for analyzing the OPA-NAC derivatives was used (A = 50 mM sodium acetate, pH 5.9, and B = methanol): 10–20% B over 3.2 min, 20% B for 1.2 min, 20–40% B over 3.6 min, 40% B for 1.2 min, 40–60% B over 3.8 min, 60% B for 1 min, and 60–10% B over 0.01 min. The calibration curve was prepared with five concentrations (10, 25, 50, 100, and 250 μM in 0.05 M HCl) of Hse and the peak heights were used for quantification.
Results and discussion
Molecular and catalytic properties of T. maritima AK-HseDH
The enzyme was purified from the crude extract in four successive steps: heat treatment followed by three column chromatography steps (). About 0.8 mg of purified enzyme was obtained from 2.0 L of E. coli culture. The purified enzyme showed a Vmax value of about 66.5 ± 0.49 μmol⋅min−1⋅mg−1 at 55°C for Hse oxidation. Moreover, we found that the purified enzyme also exhibits substantial activity for AK reaction (β-Ap production) (Vmax: 103 ± 0.52 μmol⋅min−1⋅mg−1 at 55°C). The Vmax values for the two activities are much higher than those determined for E. coli AK-HseDH at 30°C () [Citation25]. Both the AK and HseDH activities indicate that the T. maritima AK-HseDH is a novel type of AK-HseDH, in which the orientation of the AK, regulatory, and HseDH domains is totally different from those observed in the previously known AK-HseDHs.
Table 1. Purification of T. maritima AK-HseDH.
Table 2. Kinetic parameters for T. maritima, E. coli, and A. thaliana AK-HseDHs.
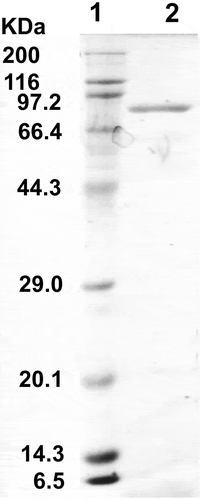
SDS-PAGE showed the subunit molecular mass of T. maritima AK-HseDH to be about 81 kDa (), which is consistent with the molecular weight (81,433) calculated from the amino acid sequence. The native molecular mass of about 450 kDa determined by gel filtration suggests the native enzyme is a homopentamer or homohexamer. In the case of A. thaliana AK-HseDH, the native molecular mass has been determined to be about 470 kDa by gel filtration, although the enzyme behaves as a tetramer (320 kDa) in the presence of 5 mM L-threonine [Citation26]. This suggests that binding of L-threonine induces conformational modification of A. thaliana AK-HseDH. On the other hand, when we carried out gel filtration of T. maritima AK-HseDH in the presence of 5 mM L-threonine, the enzyme was eluted with an apparent molecular mass of 480 kDa. The value is not substantially different from that (450 kDa) determined by gel filtration without L-threonine, indicating that the quaternary structure of this enzyme is not affected by L-threonine.
Figure 3. SDS-PAGE of recombinant T. maritima AK-HseDH. Lane 1, marker proteins; lane 2, purified enzyme.
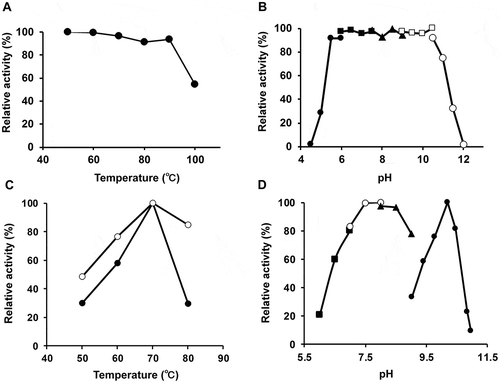
T. maritima AK-HseDH was much more stable than E. coli AK-HseDH and retained almost full activity when incubated for 10 min at temperatures up to 70°C ()), whereas E. coli AK-HseDH is rapidly inactivated above 40°C [Citation25]. The enzyme was also stable over a wide range of pH values, losing no activity when incubated at pH values between 5.5 and 10.5 for 10 min at 50°C ()). When the effect of temperature on enzyme activity was tested, we observed maximum activity at 70°C for both Hse oxidation and β-Ap production under used conditions ()). However, valid assays may not be carried out at temperatures above 70°C due to instability of the coenzymes (e.g. NADP, ATP). Evaluating the catalytic activity at different pH values revealed the enzyme to be maximally active at around pH 10.2 for Hse oxidation and pH 7.5 for β-Ap production ()). T. maritima AK-HseDH showed typical Michaelis-Menten kinetics for both the reactions at 55°C; the Km values for NADP and Hse were 0.064 ± 0.0040 and 1.81 ± 0.017 mM, respectively, and those for L-aspartate and ATP were 4.66 ± 0.16 and 3.54 ± 0.31 mM, respectively () [Citation26].
Figure 4. Thermal/pH stability and optimal pH level and temperature. (A) Effect of temperature on the stability of AK-HseDH. The enzyme was incubated for 10 min at different temperatures, and the residual activity was measured using the standard assay method for HseDH. (B) Effect of pH on the stability of AK-HseDH. The enzyme was incubated in different pH buffers for 10 min at 50°C, and the remaining activity was determined under the standard assay conditions for HseDH. (C) Effect of temperature on HseDH activity (![]()
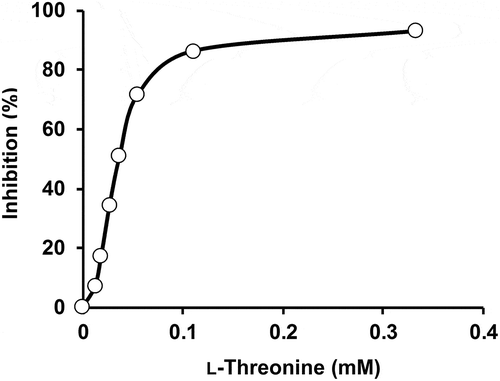
Effect of L-threonine on activities of T. maritima AK-HseDH
When the effect of L-threonine on AK activity of T. maritima AK-HseDH was examined, we observed that L-threonine inhibits β-Ap production in a cooperative manner (), similar to the case of A. thaliana AK-HseDH [Citation13]. However, the concentration of L-threonine required to inhibit the AK activity of T. maritima AK-HseDH (K0.5 = 37 μM; nH = 2.44) was much lower than that needed for inhibiting A. thaliana AK-HseDH (K0.5 = 500 μM; nH = 1.95) [Citation13]. L-Threonine also inhibits Hse oxidation of A. thaliana AK-HseDH (K0.5 = 60 mM) [Citation13]. In contrast, almost no inhibition was observed for Hse oxidation of T. maritima AK-HseDH (the relative activity with 50 mM L-threonine was 92.4% of that observed without L-threonine). These results suggest the mechanism of L-threonine-dependent inhibition differs markedly between T. maritima AK-HseDH and A. thaliana AK-HseDH. Some monofunctional AKs have been known to be markedly inhibited by L-lysine. For example, the K0.5 values for L-lysine inhibition for AK1, AK2, and AK3 from A. thaliana are 0.57 mM, 10 μM, and 7.4 μM, respectively [Citation27], and that for AKIII from E. coli is 0.5 mM. [Citation25] Thus, we examined the effect of L-lysine on both the AK and HseDH activities of T. maritima AK-HseDH, and found that the relative activities by the addition of 5 mM L-lysine were 89.5% and 93.6% of those observed without L-lysine addition. This indicates T. maritima AK-HseDH is likely to be L-lysine insensitive.
Figure 5. Effect of L-threonine on AK activity of T. maritima AK-HseDH. The data were fitted with a Hill equation (K0.5 = 37 μM; nH = 2.44).
In L-threonine-sensitive AK-HseDH, the intermediate linker domain between the AK and HseDH catalytic domains is responsible for the regulatory effect by L-threonine () [Citation15–Citation19]. In the intermediate domain of A. thaliana AK-HseDH, two potential L-threonine-binding sites have been identified [Citation13]. Based on the site-directed mutagenesis and kinetic analysis, it has been proposed that L-threonine interaction with Gln443 leads to inhibition of AK activity and facilitates the binding of a second L-threonine on Gln524. The binding of this second L-threonine leads to the inhibition of HseDH activity [Citation13].
The amino acid sequence of the regulatory domain of A. thaliana AK-HseDH (342–588) (the sequence information (AT4G19710) is available in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database) was aligned with those of E. coli AK-HseDH (255–496) (P423_00005 in KEGG) and T. maritima AK-HseDH (538–739) ()). The regulatory domain of A. thaliana AK-HseDH shares 36% sequence identity with that of E. coli AK-HseDH. In the corresponding domain of T. maritima AK-HseDH, on the other hand, 32 residues of the C-terminal part of the regulatory domains of A. thaliana and E. coli AK-HseDHs are not present ()). Both the residues Gln443 and Gln524 in A. thaliana AK-HseDH, which are proposed to interact with L-threonine, are completely conserved as Gln351 and Gln432, respectively, in E. coli AK-HseDH. However, Gln524 in A. thaliana AK-HseDH is replaced by Ala709 in T. maritima AK-HseDH, although Gln443 was conserved as Gln631. Indeed, it has been reported that the Q524A mutation of A. thaliana AK-HseDH greatly increases the K0.5 value for inhibition of Hse oxidation by L-threonine (K0.5 = 335 mM) but does not modify the K0.5 value and cooperative manner for inhibition of AK activity [Citation13]. To obtain structural information about the sites where L-threonine potentially binds, we predicted the secondary structure of the regulatory domains in A. thaliana and T. maritima AK-HseDHs using SWISS-MODEL. [Citation28] As shown in ), the two ACT subdomains (two loop-α-loop-β-loop-β motifs) were predicted for the residues 606–645 and 685–718 of T. maritima AK-HseDH, as well as for the residues 416–453 and 500–534 of A. thaliana AK-HseDH. These observations suggest that, although the two enzymes share the structurally similar ACT subdomains, the L-threonine-binding mode in T. maritima AK-HseDH is markedly different from that proposed for A. thaliana AK-HseDH, which contributes to the distinct manner of the L-threonine-dependent inhibition observed for T. maritima AK-HseDH. Therefore, it is of interest to analyze the structural difference of the regulatory domain between the two enzymes based on crystal structures.
Figure 6. Comparison of regulatory domains. (A) Amino acid sequence alignment of the regulatory domains of A. thaliana, E. coli, and T. maritima AK-HseDHs. At, A. thaliana; Ec, E. coli; and Tm, T. maritima. Asterisks indicate conserved residues. Gln residues proposed to interact with L-threonine are in boxes. The residues corresponding to the two ACT subdomains of A. thaliana (416–453 and 500–534) and T. maritima (606–645 and 685–717) enzymes are shaded. Sequences were aligned using ClustalW [Citation30]. (B) Predicted structures of the two ACT subdomains of A. thaliana (left panel) and T. maritima (right panel) enzymes. Amino acid residues (Gln443 and Gln524 for A. thaliana, Gln631 and Ala709 for T. maritima) are shown as stick models.
![Figure 6. Comparison of regulatory domains. (A) Amino acid sequence alignment of the regulatory domains of A. thaliana, E. coli, and T. maritima AK-HseDHs. At, A. thaliana; Ec, E. coli; and Tm, T. maritima. Asterisks indicate conserved residues. Gln residues proposed to interact with L-threonine are in boxes. The residues corresponding to the two ACT subdomains of A. thaliana (416–453 and 500–534) and T. maritima (606–645 and 685–717) enzymes are shaded. Sequences were aligned using ClustalW [Citation30]. (B) Predicted structures of the two ACT subdomains of A. thaliana (left panel) and T. maritima (right panel) enzymes. Amino acid residues (Gln443 and Gln524 for A. thaliana, Gln631 and Ala709 for T. maritima) are shown as stick models.](/cms/asset/783d179f-2190-4023-bb6f-ffb9ce55a095/tbbb_a_1511365_f0006_b.gif)
Overall conversion of L-aspartate to Hse
AK-HseDH is an unusual bifunctional enzyme in that it does not catalyze consecutive reactions. In the biosynthetic pathway from L-aspartate to Hse, AsaDH utilizes the product of AK (β-Ap) to form the substrate of HseDH (Asa) (). To confirm whether T. maritima AK-HseDH has the function as a part of this pathway, the overall conversion of L-aspartate to Hse was examined in the presence of T. maritima AsaDH and AK-HseDH.
To determine the stoichiometry of the two enzymes, different amounts of T. maritima AsaDH (13.2–198 pmol) were incubated with a fixed amount of T. maritima AK-HseDH (63.4 pmol), and the reaction was followed by measuring the oxidation of NADPH catalyzed by AsaDH and HseDH. As shown in , the maximum rate of NADPH oxidation was reached at the ratio of AK-HseDH : AsaDH = 1 : 1. In the case of A. thaliana AsaDH and AK-HseDH, the maximum activity was reached with non-stoichiometric amounts of AK-HseDH and AsaDH (AK-HseDH : AsaDH = 5 : 1) () [Citation29]. The oxidation rate of NADPH in the presence of T. maritima AsaDH and AK-HseDH is rather low. AsaDH may be a rate-limiting step in the overall conversion of L-aspartate to Hse because of its low reaction rate. However, further detailed investigation is required to prove this hypothesis.
Table 3. Stoichiometry of AK-HseDH and AsaDH.
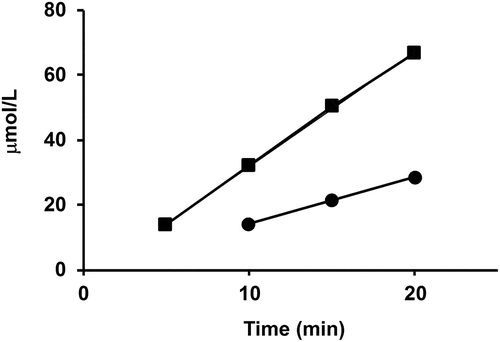
In the presence of equimolar amounts of T. maritima AK-HseDH and AsaDH, the amount of Hse produced at appropriate time intervals was determined using a UPLC system. Concomitantly, the oxidation of NADPH catalyzed by AsaDH and HseDH was monitored. As a result, we observed that the rate of NADPH oxidation was 2-times higher than that of Hse production (). This result coincides with the predicted stoichiometry in the biosynthetic pathway from L-aspartate to Hse; 2 mol of NADPH is oxidized to produce 1 mol of Hse. These results suggest that the metabolic intermediate is effectively transferred from AK to HseDH via AsaDH in a combination of T. maritima AK-HseDH and AsaDH.
Figure 7. NADPH oxidation and Hse production in the overall reaction of T. maritima AsaDH and AK-HseDH. In the presence of equimolar amounts of T. maritima AK-HseDH and AsaDH, the amount of NADPH oxidized (■) and that of Hse produced (![]()
In the present study, we first showed the presence of a novel type of L-threonine-sensitive AK-HseDH in the hyperthermophilic bacterium, T. maritima. Furthermore, the L-threonine-inhibition mechanism of T. maritima AK-HseDH was notably different from that of A. thaliana AK-HseDH. The unique order of the HseDH, AK, and regulatory domains in T. maritima AK-HseDH, as well as the distinctive amino acid sequence of the regulatory domain, is likely responsible for this difference. The AK-HseDHs with similar domain arrangement exist only in the several species of both Thermotoga and Mesotoga genera within the order Thermotogales. Our next aim is to shed light on the 3D-structure of the novel AK-HseDH and to obtain more detailed information about the L-threonine-dependent inhibition mechanism.
Notes on contributions
Haruhiko Sakuraba designed the research. Tatsuya Ohshida, Kohei Koba, Kazunari Yoneda, and Taketo Ohmori performed the experiments. Haruhiko Sakuraba, Tatsuya Ohshida, Kohei Koba, and Junji Hayashi analyzed the data. Toshihisa Ohshima and Tatsuya Ohshida participated in the review of the analysis results. Haruhiko Sakuraba and Tatsuya Ohshida wrote the manuscript. Toshihisa Ohshima also participated in the review of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Archer JA, Solow-Cordero DE, Sinskey AJA. C-terminal deletion in Corynebacterium glutamicum homoserine dehydrogenase abolishes allosteric inhibition by L-threonine. Gene. 1991;107:53–59.
- Jacques SL, Ejim LJ, Wright GD. Homoserine dehydrogenase from Saccharomyces cerevisiae: kinetic mechanism and stereochemistry of hydride transfer. Biochim Biophys Acta. 2001;1544:42–54.
- Schroeder AC, Zhu C, Yanamadala SR, et al Threonine-insensitive homoserine dehydrogenase from soybean: genomic organization, kinetic mechanism, and in vivo activity. J Biol Chem. 2010;285:827–834.
- Sibilli L, Le Bras G, Cohen GN. Two regions of the bifunctional protein aspartokinase I- homoserine dehydrogenase I are connected by a short hinge. J Biol Chem. 1981;256:10228–10230.
- Yoshida A, Tomita T, Kuzuyama T, et al Mechanism of concerted inhibition of α2β2-type hetero-oligomeric aspartate kinase from Corynebacterium glutamicum. J Biol Chem. 2010;285:27477–27486.
- Azevedo RA, Lancien M, Lea PJ. The aspartic acid metabolic pathway, an exciting and essential pathway in plants. Amino Acids. 2006;30:143–162.
- Viola RE. The central enzymes of the aspartate family of amino acid biosynthesis. Acc Chem Res. 2001;34:339–349.
- Yamaguchi H, Uchida K, Hiratani T, et al RI-331, a new antifungal antibiotic. Ann N Y Acad Sci. 1988;544:188–190.
- Galili G, Hofgen R. Metabolic engineering of amino acids and storage proteins in plants. Metab Eng. 2002;4:3–11.
- Hacham Y, Matityahu I, Schuster G, et al Overexpression of mutated forms of aspartate kinase and cystathionine gamma-synthase in tobacco leaves resulted in the high accumulation of methionine and threonine. Plant J. 2008;54:260–271.
- Krishnan HB. Engineering soybean for enhanced sulfur amino acid content. Crop Sci. 2005;45:454–461.
- Shaul O, Galili G. Concerted regulation of lysine and threonine synthesis in tobacco plants expressing bacterial feedback-insensitive aspartate kinase and dihydrodipicolinate synthase. Plant Mol Biol. 1993;23:759–768.
- Paris S, Viemon C, Curien G, et al Mechanism of control of Arabidopsis thaliana aspartate kinase-homoserine dehydrogenase by threonine. J Biol Chem. 2003;278:5361–5366.
- Umbarger HE. Amino acid biosynthesis and its regulation. Annu Rev Biochem. 1978;47:533–606.
- Fazel A, Guillou Y, Cohen GN. A hybrid proteolytic fragment of Escherichia coli aspartokinase I-homoserine dehydrogenase I. Structure, inhibition pattern, dissociation properties, and generation of two homodimers. J Biol Chem. 1983;258:13570–13574.
- Fazel A, Muller K, Le Bras G, et al A triglobular model for the polypeptide chain of aspartokinase I-homoserine dehydrogenase I of Escherichia coli. Biochemistry. 1983;22:158–165.
- Ferrara P, Duchange N, Zakin MM, et al Internal homologies in the two aspartokinase-homoserine dehydrogenases of Escherichia coli K-12. Proc Natl Acad Sci USA. 1984;81:3019–3023.
- Omori K, Imai Y, Suzuki S, et al Nucleotide sequence of the Serratia marcescens threonine operon and analysis of the threonine operon mutations which alter feedback inhibition of both aspartokinase I and homoserine dehydrogenase I. J Bacteriol. 1993;175:785–794.
- Omori K, Komatsubara S. Role of serine 352 in the allosteric response of Serratia marcescens aspartokinase I-homoserine dehydrogenase I analyzed by using site-directed mutagenesis. J Bacteriol. 1993;175:959–965.
- Chipman DM, Shaanan B. The ACT domain family. Curr Opin Struct Biol. 2001;11:694–700.
- Lo CC, Bonner CA, Xie G, et al Cohesion group approach for evolutionary analysis of aspartokinase, an enzyme that feeds a branched network of many biochemical pathways. Microbiol Mol Biol Rev. 2009;73:594–651.
- Ohshima T, Misono H, Soda K. Properties of crystalline leucine dehydrogenase from Bacillus sphaericus. J Biol Chem. 1978;253:5719–5725.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685.
- James CL, Viola RE. Production and characterization of bifunctional enzymes. Domain swapping to produce new bifunctional enzymes in the aspartate pathway. Biochemistry. 2002;41:3720–3725.
- Paris S, Wessel PM, Dumas R. Overproduction, purification, and characterization of recombinant bifunctional threonine-sensitive aspartate kinase-homoserine dehydrogenase from Arabidopsis thaliana. Protein Expr Purif. 2002;24:105–110.
- Curien G, Laurencin M, Robert-Genthon M, et al Allosteric monofunctional aspartate kinases from Arabidopsis. FEBS J. 2007;274:164–176.
- Waterhouse A, Bertoni M, Bienert S, et al SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303.
- Paris S, Wessel PM, Dumas R. Overproduction, purification, and characterization of recombinant aspartate semialdehyde dehydrogenase from Arabidopsis thaliana. Protein Expr Purif. 2002;24:99–104.
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680.