
ABSTRACT
The total synthesis of peramine, a natural product isolated from an endophytic fungi, has been achieved in four steps and 34% overall yield from known compounds. The key step was the one-pot construction of the pyrrolopyrazinone ring from pyrrole amide and propargyl bromide. The preparation of peramine-d4 as an internal standard for quantitative analysis by MS is also described.
Graphical Abstract
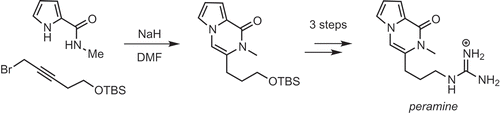
Peramine, a natural product isolated from an endophytic fungi, was synthesized in four steps by one-pot construction of the pyrrolopyrazinone ring as a key step.
Fungal endophytes infected in perennial ryegrass (Lolium perenne L.) produce a variety of structurally complex secondary metabolites [Citation1], including peramine (1), in , which was isolated by Rowan and co-workers in 1986 [Citation2,Citation3].
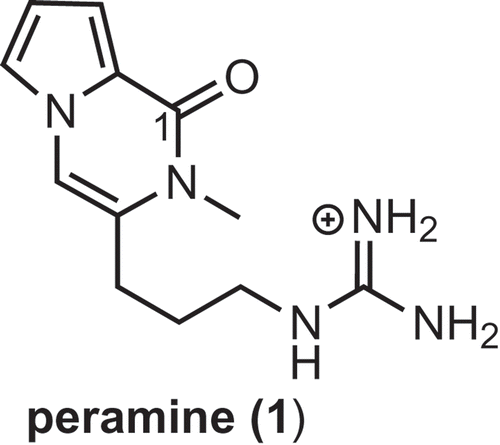
Figure 1. Structure of peramine (1).
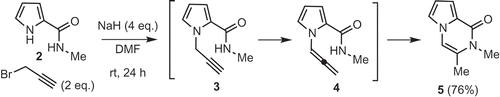
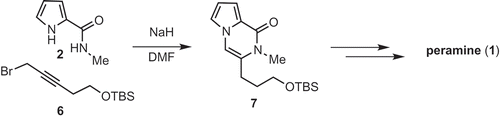
Several syntheses of peramine have been reported [Citation4–Citation7] Our interest in peramine is due to its poorly characterized biological activity and unique chemical structure – a pyrrolopyrazinone ring, virtually unknown in nature; and a guanidine moiety, our interest in which is ongoing [Citation8,Citation9]. In 2017, we reported a novel, copper-catalyzed annulation reaction between pyrrole amide and 1-bromoacetylene, as well as the synthesis of peramine from the annulation product [Citation10]. However, a preparative scale synthesis of peramine using this method proved impractical, due to the intractable nature of the product mixture obtained. Recently Balci reported a one-pot reaction of pyrrole amide 2 and propargyl bromide with sodium hydride as a base to give pyrrolopyrazinone 5 in a good yield [Citation11]. The mechanism of this reaction was also proposed to entail N-propargylation of the pyrrole to give 3, followed by isomerization to allene 4 and 6-exo-cyclization. We sought to use this one-pot reaction to synthesize key intermediate 7, which was transformed to peramine in three steps by Scheerer [Citation7] and us [Citation10] . Pyrrole amide 2 and propargyl bromide 6 are easily prepared from 2-(trichloroacetyl)pyrrole and 3-buytyn-1-ol [Citation11,Citation12], respectively.
Scheme 1. Synthesis of pyrrolopyrazinone reported by Balci.
Scheme 2. A synthetic plan of peramine (1).
Results and discussion
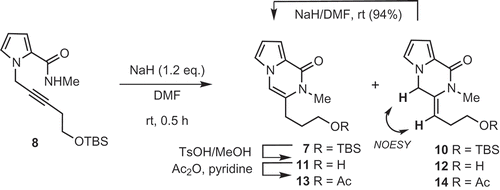
According to the procedure reported by Balci [Citation11], we first attempted to react pyrrole amide 2 and propargyl bromide 6 (2 equivalents) in the presence of sodium hydride (2 equivalents) for two hours (entry 1; ). N-Propargyl product 8 and di-N-propargyl product 9 were obtained, but no desired product 7. In order to suppress the formation of di-N-propargyl product 9, the reaction was conducted with only 1.1 equivalents of bromide 6 (entry 2). The desired pyrrolopyrazinone 7 was now obtained in a yield of 33% yield; di-N-propargyl product 9 was still formed, and unreacted starting material 2 recovered. These results indicate that cyclization of 8 is competitive to the second N-propargylation of 8 under these conditions, using an excess amount of the base. Since the same reaction with 1.2 equivalents of NaH gave a good yield of 8 (entry 3), and isolated sample of product 8 could be treated with NaH (1.2 equivalents) at room temperature for 30 min to provide 7 in 96% yield, we elected to conduct the two reactions in one-pot manner. Pyrrole amide 2 and propargyl bromide 6 were first reacted with 1.2 equivalents of NaH, and then 1.2 equivalents of NaH was added after consumption of 2. Product 7 was obtained in a yield of 67% on a gram scale, without problems (entry 4).
Table 1. Attempted synthesis of pyrrolopyrazinone 7.
The mechanism of the cyclization of 8 to give 7 with sodium hydride merits further discussion, informed by our optimization of this reaction . When the reaction was quenched after a short time (ca. 0.5 hours), exo-product 10 was also formed as a minor product, along with pyrrolopyrazinone 7 . The regiochemistry of the exo-product 10 was determined to be Z by analysis of NOESY spectra of the corresponding acetate 14, obtained by deprotection of TBS (cat.TsOH in MeOH) followed by acetylation (Ac2O, pyridine). The exo product 10 was easily isomerized to pyrrolopyrazinone 7 using NaH in DMF [Citation13].
Scheme 3. Base-promoted cyclization of 8.
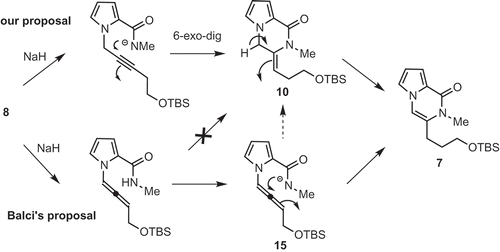
Balci reported a DFT calculation in support of the mechanism depicted in [Citation11]. However, the formation of the exo-product 10 led us to propose an alternative mechanism for the formation of pyrrolopyrazinone 7 from 8 different to that proposed by Balci; wherein direct 6-exo-dig cyclization of 8 provides exo-product 10, which undergoes isomerization to give endo product 7 (pyrrolopyrazinone). Formation of exo-product 10 from allene 15 is considered to be difficult due to the poor orbital overlap between the nucleophilic amide nitrogen and the allene π*-orbital; as well as the E nature of the double bond of 10. However, even when the reaction was quenched after a very short reaction time (4 min), a 1:1:1.2 mixture of 7, 8 and 10 was detected by 1H-NMR spectra of the crude mixture. These results suggest that both mechanisms may operate under these conditions.
Scheme 4. A newly proposed mechanism from 8 to 7.
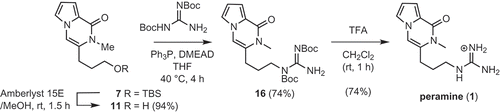
Product 7 was transformed to peramine (1) in three steps as shown in . In our previous synthesis of peramine [Citation10], the TBS group of 7 was deprotected with TBAF in THF; however, upon scaling the reaction up, this method proved problematic due to the high polarity of the product combined with the need to remove the tetra-n-butylammonium salt during work-up. Accordingly, we elected to use the acidic resin Amberlyst 15E in methanol instead. Guanidinylation of the product 11 and deprotection of the two Boc groups were carried out as previously reported [Citation7,Citation10], to furnish peramine (1) in a yield of 55% over the two steps.
Scheme 5. Synthesis of peramine (1) from 7.
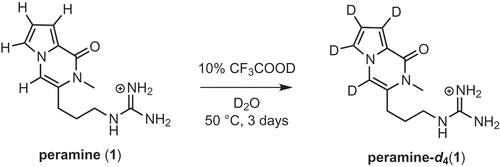
When the 1H-NMR spectra of the synthesized peramine was acquired in D2O, partial deuteration of the pyrrolopyrazinone nucleus was unexpectedly observed. Complete deuteration of the four protons of peramine was carried out by treatment with CF3COOD in D2O at 50°C for 3 days . Peramine-d4 could be useful as an internal standard of the trace analysis by GC-MS and/or LC-MS, although currently homoperamine is being used as a standard for LC-MS/MS [Citation14].
Scheme 6. Deuteration of peramine.
Conclusion
Peramine has been successfully synthesized in four steps and 34% yield from two known compounds; the one-pot construction of the pyrrolopyrazinone ring from 2 and 6 is the key reaction, for which a new mechanism is proposed based on the experimental results obtained. The resulting peramine was easily deuterated by treatment with CF3COOD in D2O to provide peramine-d4, which could be used as an internal standard of MS analysis.
Experimental
General
Infrared spectra (IR) were recorded on a JASCO FT/IR-4100 spectrophotometer, and are reported in wave number (cm−1). Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker ARX-400 (400 MHz) or ARX-600 (600 MHz) spectrometer. Chemical shifts of all compounds are reported in ppm relative to the residual standard solvent (chloroform-d as δ = 7.26, methanol-d4 as δ = 3.31). CH3CN (δ = 2.06) was used as an internal standard when 1H NMR spectrum of peramine-d4 in D2O was measured. Data are reported as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet, br = broadened), and assignment. Carbon nuclear magnetic resonance (13C NMR) spectra were recorded on a Bruker ARX-400 (100 MHz) or ARX-600 (150 MHz) spectrometer. Chemical shifts of all compounds are reported in ppm relative to the residual standard solvent (chloroform-d as δ = 77.16, CD3OD-d4 as δ = 49.0). High resolution mass spectra (HRMS) were recorded on an Applied Biosystems Mariner ESI-TOF spectrometer and reported in m/z. Reactions were monitored by TLC on 0.25 mm silica gel coated glass plates 60F254 (Merck, # 1.05715) and visualized using UV light (254 nm) and/or the developing agents 7% phosphomolybdic acid and p-anisaldehyde solution in H2SO4/AcOH/EtOH, with heat. Silica gel 60N (spherical, particle size 0.04–0.05 mm, Kanto, # 37562-84) was used for flash-column chromatography. Dry DMF was distilled from calcium hydride and stored over 4Å molecular sieves. Dry THF was purchased from Kanto Chemical Co., Inc. All other reagents were commercially available and used without further purification.
General experimental procedure of table1 exemplified by entry 1
Pyrrole amide 2 (105 mg, 0.846 mmol) was added to a suspension of NaH (ca. 60%, 67.6 mg, 1.69 mmol) in DMF (7.0 mL). After stirring at rt for 30 min, compound 6 (0.46 mL, 1.69 mmol) was added dropwise over 5 min. Stirring was continued at rt for 2 h, and then the reaction was diluted with water (3 mL). The mixture was extracted with EtOAc (x9), and the combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography (neutral silica gel, 20 g, hexane:EtOAc = 9:1 (Rf = 0.16)) to give compound 8 (84.3 mg, 31%) as pale yellow crystals and a mixture containing compound 9, which was further purified by flash column chromatography (neutral silica gel, 10 g, EtOAc) to give 9 (312 mg, 71%) as a pale yellow oil. Compound 8: 1H NMR (400 MHz, CDCl3) δ: 0.05 (6H, s, -Si(CH3)2), 0.88 (9H, s, -(CH3)3), 2.42 (2H, tt, J = 7.0, 2.0 Hz, -CH2-), 2.90 (3H, d, J = 5.0 Hz, -NCH3), 3.71 (2H, t, J = 7.0 Hz, -CH2-), 5.19 (2H, t, J = 2.5 Hz, -CH2-), 5.98 (1H, brs, -NH-), 6.09 (1H, dd, J = 4.0, 3.0 Hz, pyrrole), 6.52 (1H, dd, J = 4.5, 2.0 Hz, pyrrole), 7.10 (1H, t, J = 2.0 Hz, pyrrole). 13C NMR (100 MHz, CDCl3) δ: -5.2, 18.4, 23.2, 25.9, 26.1, 38.4, 61.8, 76.0, 83.1, 107.5, 112.0, 125.1, 126.1, 162.6. IR (KBr) vmax 3347, 2954, 2856, 1633, 1552, 1257, 1106, 837 cm−1. HRMS (ESI, positive): calcd for C17H29N2O2Si (M + H), 321.1993; found, 321.2020. Compound 9: 1H NMR (400 MHz, CDCl3) δ: 0.05 (6H, s, -Si(CH3)2), 0.07 (6H, s, -Si(CH3)2), 0.88 (9H, s, -(CH3)3), 0.90 (9H, s, -(CH3)3), 2.38–2.47 (4H, m, -CH2- x2), 3.15 (3H, brs, -NCH3), 3.70 (2H, t, J = 7.0 Hz, -CH2-), 3.73 (2H, t, J = 7.0 Hz, -CH2-), 4.28 (2H, t, J = 2.0 Hz, -CH2-), 4.95 (2H, t, J = 2.0 Hz, -CH2-), 6.11 (1H, dd, J = 4.0, 3.0 Hz, pyrrole), 6.59 (1H, brs, pyrrole), 7.04 (1H, dd, J = 2.5, 1.5 Hz, pyrrole). 13C NMR (100 MHz, CDCl3) δ: -5.16, -5.15, 18.4, 23.3, 26.0, 38.2, 61.9, 62.0, 76.1, 76.2, 82.9, 107.3, 114.0, 124.2, 125.2, 163.6. IR (KBr) vmax 2955, 2930, 2856, 1627, 1471, 1464, 1253, 1106, 836 cm−1. HRMS (ESI, positive): cald for C28H48N2O3NaSi2 (M+ Na), 539.30757; found, 539.30957.
1-(5-((tert-Butyldimethylsilyl)oxy)pent-2-yn-1-yl)-N-methyl-1H-pyrrole-2-carboxamide (8) (entry 3 of )
Pyrrole amide 2 (200 mg, 1.61 mmol) was added to a suspension of NaH (ca. 60%, 77.0 mg, 1.93 mmol) in DMF (7.0 mL). After stirring at rt for 5 min, compound 6 (0.45 mL, 1.69 mmol) was added dropwise over 5 min. Stirring was continued at rt for 4 h, and then the reaction was diluted with water (3 mL). The mixture was extracted with EtOAc (x9), and the combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography (neutral silica gel, 18 g, hexane:EtOAc = 3:1 (Rf = 0.20)) to give 8 (443 mg, 86%) as pale yellow crystals.
Synthesis of 3-(3-((tert-Butyldimethylsilyl)oxy)propyl)-2-methylpyrrolo [1,2-a] pyrazin-1 (2H)-one (7) from 8
NaH (ca. 60%, 10.8 mg, 270 µmol) was added to a solution of 8 (72.0 mg, 225 µmol) in DMF (2.0 mL). After being stirred at rt for 6 h, the reaction was diluted with water (2 mL) and extracted with EtOAc (X9). The combined organic layer was washed with brine (X2), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography (neutral silica gel, 3 g, hexane:EtOAc = 2:1) to give 7 (68.9 mg, 96%) as pale yellow crystals.
One-pot synthesis of 3-(3-((tert-butyldimethylsilyl)oxy)propyl)- 2-methylpyrrolo [1,2-a] pyrazin-1(2H)-one (7) (entry 4 of )
Pyrrole amide 2 (1.00 g, 8.06 mmol) was added to a suspension of NaH (ca. 60%, 386 mg, 9.67 mmol) in DMF (35.0 mL). After stirring at rt for 30 min, bromide 6 (2.70 mL, 8.46 mmol) was added dropwise over 5 min. Stirring was continued at rt for 3.5 h and consumption of 2 was monitored by TLC (hexane/Et2O = 1:1 (×3)). NaH (ca. 60%, 386 mg, 9.67 mmol) was then added and stirring was further continued at rt for 3.5 h. The reaction was diluted with water (15 mL) and the mixture was extracted with EtOAc (×9). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated. The residue was purified by flash column chromatography (neutral silica gel, 70 g, hexane:EtOAc = 3:1 (Rf = 0.20)) to give 7 (1.72 g, 67%) as pale yellow crystals.
Synthesis of acetates of 7 and 10
(i) TsOH•H2O (8.9 mg, 47 µmol) was added to a solution of a 3:1 mixture of 7 and 10 (128 mg) in MeOH (5.0 mL). After being stirred at rt for 15 min, the mixture was diluted with pyridine (10 μL) and toluene and then evaporated in vacuo. The residue was purified by flash column chromatography (neutral silica gel, 2 g, CH2Cl2:acetone = 5:2 (Rf = 0.26)) to give a 3:1 mixture of 11 and 12 (62 mg). (ii) The mixture of 11 and 12 was dissolved in pyridine (4.0 mL) and Ac2O (3.0 mL). After being stirred at rt for 20 min, the mixture was diluted with toluene and then evaporated in vacuo. The residue was purified by flash column chromatography (neutral silica gel, 3 g, Et2O) to give endo 13 (64 mg) and exo 14 (25 mg). Compound 13: Mp 117–120 ºC. 1H NMR (400 MHz, CD3OD) δ: 1.96 (2H, m, -CH2-), 2.02 (3H, s, -OCOCH3), 2.67 (2H, t, J = 7.0 Hz, -CH2-), 3.46 (3H, s, -NCH3), 4.17 (2H, t, J = 6.0 Hz, -CH2-), 6.54 (1H, dd, J = 4.0, 2.5 Hz, pyrrole), 6.97 (1H, d, J = 4.0 Hz, pyrrole), 7.20 (1H, s, -CH2 = ), 7.24 (1H, t, J = 2.0 Hz, pyrrole). 13C NMR (100 MHz, CD3OD) δ: 20.8, 28.0, 28.5, 29.7, 64.6, 108.4, 110.5, 113.5, 119.9, 123.7, 129.3, 159.3, 172.9. Anal. calcd for C13H16N2O3: C, 62.89; H, 6.50; N, 11.28. Found: C, 63.15; H, 6.62; N, 11.00. Compound 14: 1H NMR (400 MHz, CD3OD) δ: 1.98 (3H, s, -OCOCH3), 2.59 (2H, quartet, J = 7.0 Hz, -CH2-), 3.35 (3H, s, NCH3), 4.14 (2H, t, J = 6.5 Hz, -CH2-O-), 4.66 (2H, s, -CH2-), 5.22 (1H, t, J = 7.5 Hz, = CH-), 6.20 (1H, dd, J = 4.0, 2.5 Hz, pyrrole), 6.81 (1H, dd, J = 4.0, 1.5 Hz, pyrrole), 6.89 (1H, brt, J = 2.0 Hz, pyrrole). 13C NMR (100 MHz, CD3OD) δ: 20.7, 28.3, 35.7, 51.1, 64.4, 110.8, 114.4, 116.4, 124.5, 124.9, 136.7, 161.3, 172.8. HRMS (ESI, positive): calcd for C13H16N2O3Na (M+ Na), 271.10313; found, 271.10531.
Isomerization of compound 10 to 7
NaH (ca. 60%, 1.1 mg) was added to a solution of a 2:1 mixture of 7 and 10 (7.3 mg) in DMF (1.0 mL). The mixture was stirred at rt for 3 h. The reaction was diluted with water (3 mL) and extracted with EtOAc (X3). The combined organic layer was washed with brine (×2), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography (neutral silica gel, 3 g, hexane:EtOAc = 2:1) to give 7 (7.2 mg, 94%) as pale yellow crystals.
Deprotection of TBS ether 7
Amberlyst 15E (particle size 0.024 mm, ORGANO, 162 mg) was added to a solution of 7 (1.30 g, 4.06 mmol) in MeOH (60 mL). The mixture was stirred at rt for 1.5 h, and then filtered through a sintered glass funnel, washed with MeOH, and concentrated in vacuo. The residue was purified by flash column chromatography (neutral silica gel, 25 g, CH2Cl2:acetone:MeOH = 15:2:1 (Rf = 0.27)) to give 11 (783 mg, 94%) as colorless crystals.
Synthesis of diBoc guanidine 16
DMEAD (504 mg, 3.24 mmol) was added to a solution of 11 (223 mg, 1.08 mmol), N,N’-bis(tert-butyloxycarbonyl)guanidine (420 mg, 1.62 mmol), PPh3 (377 mg, 2.16 mmol) in THF (20 mL). The mixture was stirred at 40 ºC for 4.0 h and extracted with EtOAc (x9). The combined organic layer was washed with H2O (×5) and brine (×1), dried over anhydrous Na2SO4, and concentrated. The residue was purified by flash column chromatography (neutral silica gel, 30 g, CH2Cl2:EtOAc = 7:1 (Rf = 0.29)) to give 16 (360 mg, 74%) as colorless crystals.
Peramine (1)
TFA (5.0 mL) was added to a solution of 16 (210 mg, 0.469 mmol) in CH2Cl2 (5.0 mL) at 0 ºC. After being stirred at r.t. for 1.0 h, the mixture was concentrated in vacuo. The residue was purified by reversed phase column chromatography (Cosmosil 75C18-OPN (nacalai tesque, # 37842–11), 10 g, H2O) to give peramine (1) (125 mg, 74%) as colorless crystals.
Deuteration of peramine (1)
A solution of peramine (1) (13.6 mg, 37.7 µmol) in 10% CF3COOD in D2O (0.75 mL) was stirred at 50 ºC for 72 h. The mixture was diluted with toluene and concentrated in vacuo. Peramine-d4: 1H NMR (400 MHz, D2O) δ: 1.76 (2H, m, -CH2-), 2.44 (2H, t, J = 7.5 Hz, -CH2-), 3.24 (2H, t, J = 6.0 Hz, -CH2-), 3.28 (3H, s, -NCH3). 13C NMR (150 MHz, CD3OD) δ: 28.0, 28.3, 29.4, 41.3, 107.9 (t, JC-D = 29 Hz, C-D), 110.0 (t, JC-D = 26 Hz, C-D), 112.9 (t, JC-D = 26 Hz, C-D), 119.2 (t, JC-D = 29 Hz, C-D), 123.4, 128.5, 158.5. IR (ATR) vmax 3453, 3378, 1785, 1676, 1219 cm−1. HRMS (ESI, positive): calcd for C12H14D4N5O (M+), 252.1757; found, 252.1747.
Supplemental_materials001.pdf
Download PDF (2 MB)Acknowledgments
The authors thank Prof. T. Kusumi (Tokyo Institute of Technology) and Mr. K. Koga (Nagoya University) for valuable discussion on 13C NMR spectrum of peramine-d4. Y.Y. thanks to the Program for Leading Graduate Schools: IGER Program in Green Natural Sciences from MEXT.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Panaccione DG, Beaulieu WT, Cook D. Bioactive alkaloids in vertically transmitted fungal endophytes. Funct Ecol. 2014;28:299−314.
- Rowan DD, Hunt MB, Gaynor DL. Peramine, a novel insect feeding deterrent from ryegrass infected with the endophyte Acremonium loliae. J Chem Soc Chem Commun. 1986;935–936.
- Rowan DD, Tapper BA. An efficient method for the isolation of peramine, an insect feeding deterrent produced by the fungus Acremonium Lolii. J Nat Prod. 1989;52:193–195.
- Dumas DJ. Total synthesis of peramine. J Org Chem. 1988;53:4650–4653.
- Brimble MA, Rowan DD. Synthesis of peramine, an insect feeding deterrent mycotoxin from Acremonium lolii. J Chem Soc Chem Commun. 1988;978–979.
- Brimble MA, Rowan DD. Synthesis of the insect feeding deterrent peramine via Michael addition of a pyrrole anion to a nitroalkene. J Chem Soc Perkin Trans. 1990;1:311–314.
- Nelli MR, Scheerer JR. Synthesis of peramine, an anti-insect defensive alkaloid produced by endophytic fungi of cool season grasses. J Nat Prod. 2016;79:1189–1192.
- Nishikawa T, Isobe M. Synthesis of tetrodotoxin, a classic but still fascinating natural product. Chem Rec. 2013;13:286–302.
- Nishikawa T, Urabe D, Adachi M, et al. Multifunctionality of the N-trichloroacetyl group developed in the synthesis of tetrodotoxin, a puffer fish toxin. Synlett. 2015;26:1930–1939.
- Yamamoto Y, Nakazaki A, Nishikawa T. New regiocontrolled syntheses of pyrrolopyrazinones and its application to the synthesis of peramine. Tetrahedron. 2017;73:3443–3451.
- Çentinkaya Y, Balci M. Selective synthesis of N-substituted pyrrolo[1,2-a]pyrazin-1(2H)-one derivatives via alkyne cyclization. Tetrahedron Lett. 2014;55:6698–6702.
- Berger D, Citarella R, Dutia M, et al. Multidrug resistance reversal agents. J Med Chem. 1999;42:2145–2161.
- The same isomerization occurred under acidic conditions (p-TsOH/MeOH, rt, 4 days, 77% yield).
- Koulman A, Lane GA, Christensen MJ, et al. Peramine and other fungal alkaloids are exuded in the guttation fluid of endophyte-infected grasses. Phytochem. 2007;68:355–360.