
ABSTRACT
DD(35)E motif in catalytic core domain (CCD) of integrase (IN) is extremely involved in retroviral integration step. Here, nine single residue mutants of feline foamy virus (FFV) IN were generated to study their effects on IN activities and on viral replication. As expected, mutations in the highly conserved D107, D164, and E200 residues abolished all IN catalytic activities (3′-end processing, strand transfer, and disintegration) as well as viral infectivity by blocking viral DNA integration into cellular DNA. However, Q165, Y191, and S195 mutants, which are located closely to DDE motif were observed to have diverse levels of enzymatic activities, compared to those of the wild type IN. Their mutant viruses produced by one-cycle transfection showed different infectivity on their natural host cells. Therefore, it is likely that effects of single residue mutation at DDE motif is critical on viral replication depending on the position of the residues.
Graphical Abstract
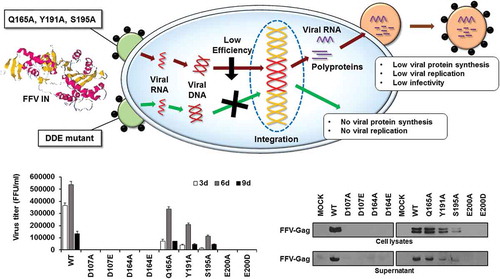
Missense mutation in feline foamy virus integrase seriously decreases production of viral progeny.
Foamy viruses (FVs) are a genus in the sub-family Spumaretrovirinae, one of the Retroviridae families [Citation1,Citation2]. FVs are the oldest retroviruses and have coevolved with their hosts for almost a billion years. The coevolution has been linked to the non-pathogenicity of FVs, a major factor in the development of foamy viral vectors in gene therapy [Citation3–Citation5]. Foamy viral vectors present some advantages such as efficient delivery of transgenes in large animal models and a probably safer integration profile. Furthermore, in many antiretroviral drug studies, FVs are used as a model virus since FV integrase (IN) has been crystallized and studied in detail [Citation6,Citation7].
For successful retroviral replication to occur, the viral cDNA must be integrated into the host cellular DNA. The integration of viral DNA is mediated by the virally encoded protein IN, which is located in the 3′ part of pol gene ()). IN mediates three enzymatic reactions in vitro, which are similar to the process mediated by polynucleotidyl transferase, viz., 3′-end processing, strand transfer and disintegration [Citation8,Citation9]. In 3′-end processing, the highly conserved CA dinucleotide from the 3′-end of linear viral DNA exposes the 3′-OH group at both ends [Citation10,Citation11]. Strand transfer is a concerted cleavage–ligation step during which IN makes a staggered cut in the chromosomal targeted DNA and ligates the recessed 3′-ends of the pre-processed viral DNA into opposing strands at the target DNA cleavage site [Citation12,Citation13]. The disintegration reaction recovers viral cDNA from the integrated DNA complexes, which is only observed in vitro (Fig. S1) [Citation14].
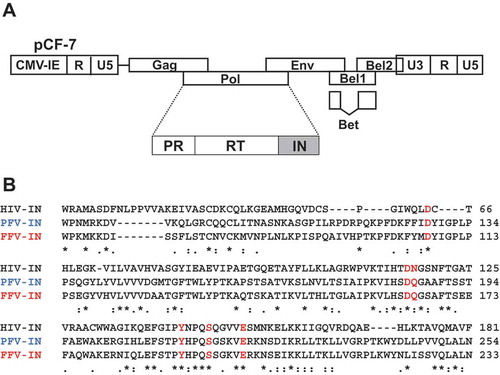
Figure 1. Design of FFV IN mutant. (a) Schematic illustration of the FFV genome based on the pro-viral clone pCF-7. (b) Amino acid alignment of CCD regions of HIV-1 IN, PFV IN, and FFV IN. HIV-1 IN sequence (upper) and FFV IN sequence (lower) were obtained from GenBank code accession numbers NC_001802 and CAD92796.1, respectively. PFV IN sequence (middle) is from protein database (PDB) code 3OY9. The six selected amino acid positions of FFV IN are marked with red letter; D107, D164, Q165, Y191, S195, and E200. The symbols denote the degree of conservation: *, identical;:, conservative; ., semi-conservative; blank, non-conservative.
The IN protein consists of three domains, as observed by the three-dimensional structure, the N-terminal domain (NTD), catalytic core domain (CCD), and C-terminal domain (CTD) [Citation15]. CCD contains the most highly conserved regions of INs and displays close structural homology to the prokaryotic enzyme transposase. The major catalytic active site of CCD is composed of three invariant acidic residues, the DD(35)E motif (Asp, Asp-35-Glu), which form a enzymatic triad with two sites that can coordinate several divalent cations (Mg2+, Mn2+, Zn2+, Cd2+, and Ca2+) [Citation16,Citation17].
The characteristics of the integration reactions and significance of the DDE motif for successful viral replication and infectivity have been well reported for human immunodeficiency virus (HIV) IN [Citation18–Citation20] and prototype foamy virus (PFV) IN [Citation21], but only a few studies on feline foamy viral (FFV) IN has been reported [Citation22]. Recent studies have revealed that simian foamy virus (SFV) is transmitted to humans who interact with infected nonhuman-primates, whereas bovine foamy virus and FFV rarely or never infect humans [Citation23,Citation24]. Further research on biochemical properties and pathogenesis of FFV would clarify the utility of FVs as gene therapy vectors and an antiretroviral drug model virus.
Here, we constructed nine single residue mutation in FFV IN CCD. In this study, we found that D107, D164, and E200 mutants exhibited almost no enzymatic activities. Thus, it may be inferred that single residue mutation in FFV IN DDE motif has adverse effects on viral integration. In addition, Q165A, Y191A, and S195A mutants exhibited reduced IN enzymatic activities and viral production compared with those of the wild-type (WT) virus. Thus, our results demonstrated that DDE motif and other near active sites of FFV IN are critical for viral protein synthesis, replication, and infectivity.
Materials and methods
Cell culture
The CRFK cell line (Korean Cell Line Bank, Seoul, Korea) was cultured and maintained in Dulbecco′s Modified Eagle′s Medium supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich, St. Louis, MO, USA), 2 mM L-glutamine, 100 μg/mL streptomycin, and 100 U/mL penicillin. The FeFAB cell lines derived from CRFK cell line containing FFV LTR-β-galactosidase reporter cassettes were maintained by adding 600 μg/mL G418.
Cloning of the FFV IN point mutant
The FFV IN expression vector was constructed by FFV IN cDNA-based PCR amplification from pCF-7 [Citation25], molecular clone DNA of FFV, and by inserting the resultant PCR products into the expression vector pQE9. The oligonucleotide primers used for construction are listed in supplemental table 1. The successful recombinant construct (pQE-FIN) was confirmed to contain 6-histidine codons upstream of the FFV IN sequences. The 6-histidines in the expressed protein afforded selective affinity for a nickel-chelated absorbent. Point mutations in the FFV IN DNA were constructed as follows. First, the DNA fragments encoding the FFV IN mutants were constructed by overlapping PCR. In the first PCR, two DNA fragments were amplified using sequence-specific primers and pQE-FIN as the template. After purification, the two PCR products were mixed at a molar ratio of 1:1 and used as the template for the second PCR. FFVIN-1Val and FFVIN-383SAPCF as primers were used for the second PCR. All PCR reactions were performed with pfu DNA polymerase (Bioneer, Korea) to avoid DNA synthesis errors. The sequences of PCR-amplified DNA fragments were verified by a commercial sequencing company (Macrogen, Seoul, Korea). The confirmed FFV IN mutants were inserted into pCF7 by restriction enzyme digestion and DNA ligation. Briefly, the nine different mutants were digested with AvaΙ and SalΙ. The isolated DNA fragments were inserted into SalΙ-defective pGEM-5zf, the intermediate vector. Finally, the confirmed intermediate vectors encoding FFV IN were cloned into pCF7 with NsiI.
Expression and purification of IN proteins
The resultant IN expression vector plasmid DNA was transformed into the E. coli strain, XL-1 blue. The transformants were proliferated at 37°C in 500 mL of LB medium containing 50 mg ampicillin. Isopropyl β-D-1 thiogalactopyranoside (0.3 mM) was added at an optical density of 0.8 to induce expression, and the culture was proliferated for additional 4 h. After harvesting, the His-tagged proteins were purified by standard procedures and stored at −80°C. Frozen bacterial pellets were thawed and resuspended in 16 mL of S1 lysis buffer [50 mM Tris-HCl (pH 7.6), 20 mM β-mercaptoethanol, 0.1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 10% glycerol, and 10 mM imidazole]. The cell suspension was kept on ice for 30 min. Then, 4 mL of 5 M NaCl and 2.2 mL of 100 mM CHAPS were added. The suspension was sonicated for 3 min on ice and centrifuged at 40,000 × g for 20 min at 4°C. The supernatant was directly loaded onto a Talon-column (bed volume of 1 mL, Clontech, Palo Alto, CA, USA) pre-equilibrated with S10 buffer [1 M NaCl and 10 mM CHAPS]. The resin was washed four times with 0.3 mL of S10 buffer. Protein was eluted ten times with 0.15 mL S100 buffer [1 M NaCl and 100 nM CHAPS]. Fractions containing the protein were collected and diluted with nine volumes of buffer A [50 mM Tris-HCl (pH 7.6), 200 mM NaCl, 1 mM dithiothreitol (DTT), 0.1 mM EDTA, and 10% glycerol] before loading onto a column containing 0.25 mL of heparin–agarose. The column was washed with 1.5 mL of buffer A. The protein was eluted twelve times with 0.2 mL of buffer C [50 mM Tris-HCl (pH 7.6), 1 mM DTT, 0.1 mM EDTA, 1 M NaCl, 10% glycerol], and stored at −80°C. Protein concentrations were determined by the Bradford method using bovine serum albumin (Bio-Rad, California, USA) as a standard.
In vitro enzymatic activity assay
The oligonucleotides used as DNA substrates for enzyme activity assays are listed in supplemental table 1. The substrates used to assay enzyme activities were double-stranded oligonucleotides containing sequences derived from the U5 LTR end of FFV DNA. The substrates were prepared by labelling the 5′-end of the sense direction oligonucleotides (FFVU5/20S, FFVU5/20S-2, and FT1) with [γ−32P] ATP and T4 polynucleotide kinase. The 3′-end processing substrate was prepared by annealing the 5′-end-labeled FFVU5/20S oligonucleotide with the complementary oligonucleotide FFVU5/20A. The substrate of the strand transfer reaction was prepared by annealing the 5′-end-labeled FFVU5/20S-2 oligonucleotide with the complementary oligonucleotide FFVU5/20A. The FFV disintegration substrate (Y-oligomer) was prepared by annealing the 5′-end-labeled FT1 oligonucleotide with FT2, FD4, and FFVU5/20A. The annealed Y-oligomer was purified on a 15% native polyacrylamide gel. In all three assays, 0.1 pmol of labeled DNA substrate was incubated with 3 pmol of IN for 60 min at 37 °C in 10 μL of reaction buffer [0.05% Nonidet P40, 0.01 mM EDTA, 20 mM HEPES (pH 7.5), 1 mM DTT, 140 mM NaCl, 1 mM CHAPS, 6 mM Tris, and 5 mM MnCl2]. The products were then separated by electrophoresis on 15% polyacrylamide gels containing 7 M urea in Tris-taurine EDTA buffer. After gel drying, the products were quantified using a Cyclone Molecular Dynamic PhosphoImager (PerkinElmer, Massachusetts, USA).
Virus production and infection test
WT and mutant viral particles were produced by transfection with pCF7 and mutant viral DNA using a polyethylenimine transfection reagent. For one-cycle viral production, we used COS-1 cells instead of CRFK cells. COS-1 cells (3 × 105) were seeded onto 60-mm culture dishes. Once the culture reached approximately 70% confluency, it was transfected with 5.8 μg of either pCF7 or mutant DNA. The transfected cells were then incubated at 37°C with 5% CO2. At 3 days post-transfection, the culture supernatants were collected by centrifugation at 22,000 × g for 15 min at 4°C, and the cell debris was discarded. The supernatant was used to measure viral titer using the FeFAB assay [Citation26], and viral pellets were prepared by additionally centrifuging the culture supernatants at 100,000 × g for 1 h at 4°C and then lysed directly with sodium dodecyl sulfate (SDS)-loading dye. Cell lysates were prepared and analyzed by western blotting using an anti FFV-Gag antibody (1:1,000 dilution). For infection experiments, CRFK cells were infected with WT virus at 0.1 MOI, which was prepared in the transfection experiment. In the infection experiments with mutant viruses, we used equal volume of the culture supernatants collected from the transfection experiments because the western blot data for the culture supernatants of the transfection experiments showed similar levels of viral Gag protein. At 3, 6 and 9 days post-infection the culture supernatants were collected and viral titers were measured by the FeFAB assay. At 6 days post-infection, cell lysates were prepared and analyzed by western blotting with an anti FFV-Gag antibody, and viral pellets were prepared by centrifuging the culture supernatants at 100,000 × g for 1 h at 4°C and then lysed directly with SDS-loading dye.
FeFAB assay
FeFAB cells are modified CRFK cells that express β-galactosidase under the control of the FFV LTR, which is transactivated by the FFV Tas protein in relation to the level of viral replication. Approximately 2 × 105 FeFAB cells were infected with the serially diluted viral solution. After 48 h, the cells were fixed with a solution [0.2% (w/v) glutaraldehyde, 1% (w/v) formaldehyde in PBS]. After washing with PBS, the fixed FeFAB cells were incubated for 4 h with an X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) staining solution. The number of blue cells were then counted using an inverted microscope. All data are representative of three independent experiments with triplicate samples.
Western blot
To detect the FFV Gag protein by western blotting, we prepared a specific anti-FFV-Gag antisera by immunizing rabbits with a synthetic peptide of 15 amino acids (GPPGPNPYRRFGDGG) representing residue 431 to residue 445 in the FFV Gag polypeptide. Uninfected and infected CRFK cells were lysed in RIPA buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1% SDS, 0.5 % (w/v) sodium deoxycholate]. Cell lysates were then centrifuged at 22,000 × g for 15 min at 4°C. After centrifugation, the clarified supernatant was collected and stored at −20°C. The protein concentration of the supernatant was measured by the Bradford assay. The culture supernatant containing viral particles was also collected by centrifuging at 100,000 × g for 1 h. The pellets were lysed directly in SDS-loading dye and stored at −80°C prior to gel electrophoresis. Equal amounts of samples were separated by 12.5% SDS-polyacrylamide gel electrophoresis (PAGE) at 120 V for 1.5 h. Proteins were then transferred to nitrocellulose membranes (GE Healthcare UK Ltd., Buckinghamshire, England) at 40 V for 1.5 h using semi-dry transfer (Hoefer, Inc., Holliston, MA, USA). The membranes were blocked for 16 h at 4°C with blocking buffer PBST [5% (w/v) non-fat dry milk, 0.1% (w/v) Tween 20 in PBS]. The membranes were then probed with the in-house rabbit polyclonal antibody (1:1,000 dilution) against FFV-Gag protein in PBST solution for 1 h. After washing with PBST, the membranes were incubated with goat anti-rabbit IgG conjugated to horseradish peroxidase (1:10,000 dilution; Sigma-Aldrich, USA) in PBST for 1 h at room temperature. The membranes were washed three times with PBST and developed using a chemiluminescence (ECL) detection kit (Bionote, Korea). As an internal control, β-actin was probed with a mouse monoclonal antibody against β-actin (1:5000 dilution, Thermo Scientific, Waltham, MA, USA) and then with goat anti mouse IgG conjugated to horseradish peroxidase.
Analysis of viral DNA integrated into chromosomal DNA
To determine whether viral DNA was integrated into cellular chromosomal DNA, CRFK cells were infected with WT virus at 0.1 MOI. For infection with the mutant viruses we used the culture supernatants of the transfection experiments. The volumes of the transfection culture supernatants used for infection were same as the volume of the WT virus, because they showed very similar levels in production of Gag protein in the western blot data of the transfection experiments. At 24 and 48 h post-infection, the cells were harvested by trypsinization. Cellular DNA extraction was performed with the Genomic DNA mini kit, following the manufacturer’s protocol (Thermo Scientific, Waltham, MA, USA). The amounts of genomic DNA were quantitated using the NanoDrop spectrophotometer. For competitive PCR, the artificial competitive template (Δstu-Ι FIN) was constructed by deleting 285 bp between two StuI sites (located at 651 and 936 nt) in the FFV IN DNA cloned into the expression vector pQE-FIN. The following oligonucleotides were used for PCR: forward primer, FFVIN-1val; reverse primer, FFVIN-383SA, as mentioned above. The WT, Q165A, Y191A, and S195A reaction mixtures (20 μL) contained 1x PCR reaction buffer, 0.5 mM dNTPs, 1 μM primers, 0.5 U of Taq polymerase, 1 μL of serial dilutions of purified Δstu-Ι FIN (117 ng/μL), and 100 ng of extracted DNA. The D107A, D164E, and E200D reaction mixtures (20 μL) contained 1x PCR reaction buffer, 0.5 mM dNTPs, 1 μM primers, 0.5 U of Taq polymerase and extracted DNA (100, 300, 500, 700 and 1000 ng, respectively). Amplification was conducted under the following conditions: 94°C for 1 min, 55°C for 1 min, 72°C for 1 min for 30 cycles. The PCR product sizes were 1,149 bp for FFV IN DNA and 864 bp for Δstu-Ι FIN DNA. PCR products were separated on 1.2% agarose gels.
Statistical analysis
All data shown in the figures are expressed as mean ± SEM. Statistical significance was analyzed with a two-tailed Student’s t test. * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
Results
In vitro enzymatic activity assay
We constructed nine single residue mutants, viz., D107A, D107E, D164A, D164E, Q165A, Y191A, S195A, E200A, and E200D ()), using overlapping PCR method [Citation27] in FFV IN CCD based on sequence alignments with HIV-1 IN and PFV IN. D107, D164, and E200 are present in the DDE motif of FFV IN. Q165, Y191, and S195 correspond to N117, Y143, and S147 in HIV-1 IN, respectively, which have been previously well-studied in antiviral-drug resistant variants, particularly for IN strand transfer inhibitors [Citation28–Citation30]. The mutated FFV INs were expressed in E. coli and consecutively purified by chromatography using Ni-NTA and heparin–agarose columns (Fig. S2) [Citation31]. The activities of IN mutants on three in vitro enzymatic reactions, 3′-end processing, strand transfer, and disintegration, were evaluated. The principles of these assays are depicted in supplemental Figure 1. The oligonucleotides used as DNA substrates for the assays are listed in supplemental table 1. Enzymatic activities of each mutant IN protein are presented as percentages of WT IN protein activity ()).
Figure 2. Significant reduction in the enzymatic activities by mutation of CCD residues in FFV IN. (a) 3′-end processing, strand transfer, and disintegration activities of mutant INs are presented as percentages of WT activity. Data are represented as mean ± SEM of three independent triplicate experiments (n = 9). The reaction products were quantitated with a Cyclone Molecular Dynamic PhosphoImager (PerkinElmer). (b) 3′-end processing reactions of mutant INs using FFV U5-mimicking 20/20mer oligonucleotide substrate. Conversion of the 20-mer oligonucleotide substrates to the 18-mer oligonucleotide products was analyzed in a 15% polyacrylamide gel. (c) Strand transfer reaction of mutant INs on FFV U5-mimicking 18/20mer oligonucleotide substrate. The strand transfer products appeared as 18 + n mer and were analyzed in a 15% polyacrylamide gel. (d) Disintegration reaction of mutant INs on Y-shaping FFV U5 oligonucleotide substrate. Production of the 30-mer oligonucleotides were analyzed in a 15% polyacrylamide gel.
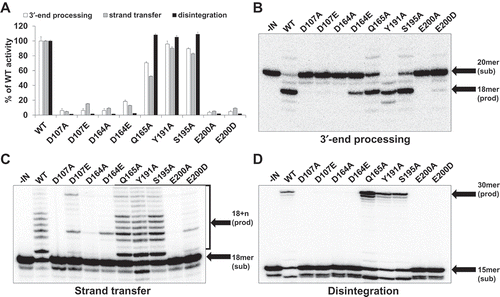
For 3′-end processing, DDE mutants exhibited less than 10% of WT activity, except for D164E IN, which exhibited 18% of WT activity. In particular, E200A exhibited only 3.7% of WT activity, and E200D exhibited the lowest activity among the three single amino acid substitutions D107E, D164E, and E200D, which were changed into related amino acids of identical charge ()). In case of strand transfer, although Q165A, Y191A, and S195A showed 52–90% of WT activity, DDE mutants presented only 4–15% of WT activity, indicating that the DDE motif is crucial for strand transfer ()) and disintegration results also revealed that DDE mutants are critical for IN protein activity. While DDE mutants lost their enzymatic activities, Q165A, Y191A, and S195A showed high activities similar to those of WT ()). The structural basis for the catalytic importance of the DDE motif has been revealed through retroviral IN X-ray crystal structures [Citation32].
According to the data shown here, any single amino acid change in FFV IN CCD exhibits decisive effects on IN catalytic effects. The D107, D164, and E200 residue position contributes to IN enzymatic activities than any other residue positions in CCD. The interesting thing is that substitution of DDE motif residue into related amino acids of identical charge is likely to be more effective for DNA cleavage and integration than substitution into alanine.
In vitro viral infectivity assay
As observed, DDE mutant IN proteins exhibited deficient catalytic activities with respect to viral DNA integration. We next determined whether viruses containing these mutations could replicated in FFV natural host cells, Crandell–Ress feline kidney (CRFK) cells. In order to study this question, first, we constructed mutant viral DNAs by introducing mutagenic DNA fragments of FFV IN into the FFV molecular clone DNA pCF-7 ()). With these viral DNAs, we separately studied viral production and viral titer in transfection and infection experiments. In transfection experiments, we used COS-1 cells instead of CRFK cells to allow only a one-cycle viral production. The COS-1 cells derived from monkey kidney tissue can produce by infectious virions through transfection with feline foamy viral DNA, but they do not allow spreading infection since these cells lack the receptor for FFV. By contrast, CRFK can produce new progenitive virions from both transfection and infection (). Therefore, the culture supernatant from CRFK transfection contains infectious virions produced not only from transfection, but also from re-infection with early produced virions. Viral titer was assessed using the FeFAB assay. Q165A, Y191A, S195A and WT viruses showed 4.6 × 104, 1.8 × 104, 4.2 × 103, and 1.1 × 105 focus forming unit (FFU) per mL of culture supernatant at 3 days post-transfection, respectively. However, D107A, D107E, D164A, D164E, E200A, and E200D mutants produced extremely low titers of virus, ranging from 120 to 210 FFU per mL of culture supernatant at 3 days post-transfection () and Fig S3A). Western blot analysis of transfected cell lysate and culture supernatant, whereby the FFV Gag proteins have been detected as 52 kDa (a full length) and 48 kDa (a cleaved product) [Citation33–Citation35], showed similar levels of Gag in WT and all nine mutant viruses ()). Although viral titer was highly different, viral particle formation was not affected by IN single residue mutations, indicating that DDE mutant virus exhibited little infection ability. Thus, we used the same volume of transfection culture supernatant to infect CRFK cells in infection experiment, whether it has infectious virions or not. From the FeFAB assay with the infection culture supernatant at 3, 6, and 9 days post-infection, blue cells were completely undetectable in DDE mutant viruses. However, substantial amounts of blue cells were detected with WT, Q165A, Y191A, and S195A infection culture supernatant until 9 days post-infection () and Fig. S3B). According to the data shown here, any substitution in the CCD of FFV IN disrupt FFV replication, especially impairment of DDE motif severely restricts spreading infection. Next, we performed western blot for 6 days post-infection cell lysates and supernatants ()). FFV Gag proteins were detected in the cell lysates and culture supernatants of WT-, Q165A-, Y191A-, and S195A-infected CRFK cells, but they were not detected in CRFK infected with DDE IN mutants. The amount of Gag proteins was related to the virus titer. Based on these data, we assumed that non-infectious virions are produced by transfection but do not infect feline host cells. Because the virions had mutated INs, they executed aberrant integration or were unable to integrate the viral DNA into the host cellular DNA and could not, thus, produce progenitive DNAs for new virion particles. The degree of viral production showed similar results to those of infectivity; WT exhibited the highest productivity, followed by Q165A, Y191A, and S195A in that order. In particular, DDE mutant viruses entirely lost their ability of viral production and infection.
Figure 3. Comparison of virus production between COS-1 cell and CRFK cell. Virus titer was detected in the supernatant of FFV transfection and infection experiments. Viral production caused by 5.8 μg DNA transfection or by an infectivity of 0.1 MOI in COS-1 and CRFK cells at 3 days post-transfection or post-infection, respectively. Data represent mean ± SEM of two independent triplicate experiments (n = 6).
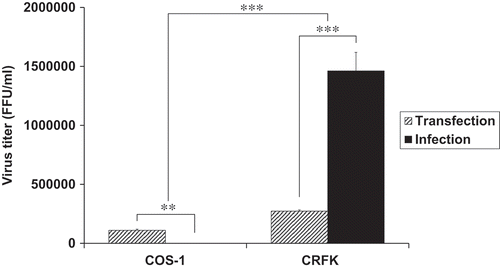
Figure 4. Viral production and infectivity by transfection. (a) Viral production in COS-1 cells by transfection. The cells were transfected with WT and mutant viral DNAs. The culture supernatants were collected at 3 days post-transfection, and virus titers were measured by the FeFAB assay. All data are representative of three independent experiments with triplicate samples in the FeFAB assay. Each bar represents mean ± SEM. (b) Western blot analysis of FFV-Gag protein in the transfected cells. Viral Gag proteins were detected with anti FFV-Gag antibody in the cell lysates and supernatant at 3 days post-transfection. β-actin as the loading control (antibody: Thermo Scientific, Waltham, MA, USA) was detected in the cell lysates.
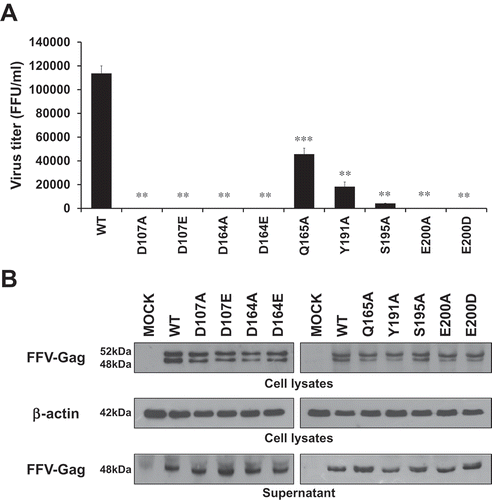
Figure 5. Viral production and infectivity by infection. (a) Viral production and infectivity study by infection. CRFK cells were infected with same volumes of the 3 days post-transfection culture supernatant of WT and each mutant virus. The culture supernatants were harvested at 3, 6, and 9 days post-infection, and virus titers were measured by the FeFAB assay. All data are representative of three independent experiments with triplicate samples in the FeFAB assay. Each bar represents mean ± SEM. (b) Western blot analysis of FFV-Gag protein in the infected cells. Viral Gag proteins were detected with anti FFV-Gag antibody from cell lysates and supernatant at 6 days post-infection.
Analysis of integrated viral DNA
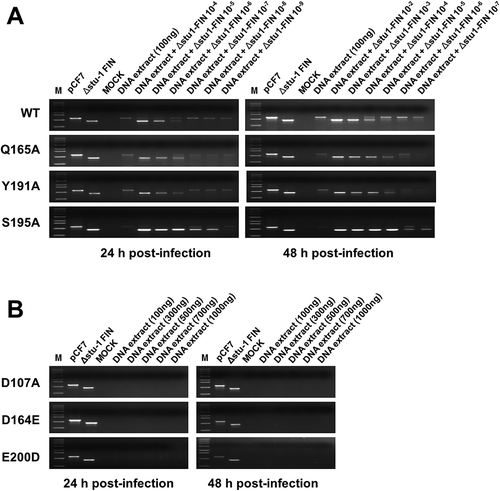
Reverse transcription of the retroviral RNA genome results in the formation of a linear double-stranded viral DNA molecule carrying a copy of the long terminal repeat (LTR) sequence at either ends. Other viral DNA remain as circular viral DNA forms, such as 1-LTRs, circular DNA with a single LTR or 2-LTRs, and circular DNA with two tandem LTRs [Citation36,Citation37]. We further sought to determine whether the decreased viral infectivity resulted from a lack or faulty integration. Therefore, we tried to detect the integrated viral DNAs from infected CRFK chromosomal DNAs and compared the extent of viral integration. For this study, first, we constructed the competitive target DNA, Δstu-Ι FIN, a plasmid containing a 285-bp fragment-deleted FFV IN DNA. Deletion of the 285-bp fragment was completed using two StuI sites in FFV IN DNA. In this competitive PCR, the 1,149-bp FFV IN region was targeted to be amplified using two oligonucleotide primers corresponding to FFV IN start and end positions, respectively. pCF-7, a plasmid encoding the FFV molecular clone, was used as the target DNA for positive control. Δstu-Ι FIN was used as a competitive PCR target DNA and was expected to yield an 864-bp DNA amplicon. CRFK cells were infected with WT virus at 0.1 multiplicity of infection (MOI) using transfection culture supernatant. Other IN mutant viruses such as D107A, D164E, Q165A, Y191A, S195A, and E200D, representing six amino acid positions, were selected and used to infect CRFK cells with transfection culture supernatants, respectively, of which volume were equal to that of WT, because western blot data for supernatants of the transfection experiments showed similar levels for Gag protein. Chromosomal DNA containing integrated viral double stranded DNA was extracted at 24 and 48 h post-infection. The chromosomal DNA isolated from the infected CRFK cells was added as the target DNA in the absence or presence of Δstu-Ι FIN in 10-fold serial dilutions. Using WT, Q165A, Y191A, and S195A samples, only the competitive DNA sample (864 bp) was amplified at a high concentration of Δstu-Ι FIN. However, as the concentration of Δstu-Ι FIN decreased, full-length (1,149 bp) FFV IN DNA was amplified, indicating that the extracted chromosomal DNA had integrated viral DNA. PCR amplicon bands switched from a 864-bp fragment to a 1,149-bp fragment when Δstu-Ι FIN in 10−5 diluted WT sample, 10−6 diluted Q165A and Y191A samples, and 10−7 diluted S195A sample at 24 h post-infection ()). These data implied that the DNA samples prepared from WT-, Q165A-, Y191A- and S195A-infected cells contained integrated viral DNA. However, compared to WT sample, Q165A, Y191A, and S195A samples had lower copies of integrated viral DNA in the same concentration of chromosomal DNA. Although these mutants were not IN DDE motif mutants, amino acid substitution in CCD affected their integration activities. The IN DDE motif mutation is critical for FFV viral integration, we could not observe amplification of the 1,149- bp fragment even at 10-fold higher amounts of chromosomal DNA ()). These results indicate that the mutant FFV IN proteins had been functionally modified so they were unable to efficiently mediate integration. In particular, DDE mutant IN proteins entirely lost their enzymatic activities and finally could not execute integration of viral DNA in the host cellular DNA.
Figure 6. Analysis of viral DNA integrated into the host chromosomal DNA. (a) Competitive PCR to detect the integrated viral DNA in the chromosomal DNA of the WT virus- and mutant virus-infected cells. CRFK cells were infected with FFV WT, Q165A, Y191A, and S195A mutant viruses by using 3 days post-transfection culture supernatants of same volume, respectively (about 0.1 MOI as WT basis). At 24 and 48 h post-infection, the infected cells were lysed. Chromosomal DNAs were isolated and quantitated. M, DNA marker (0.5, 1, 1.6, 2, 3, 4, 5, 6, 8, 10 kb from the bottom of the gel); pCF-7, pCF-7 only-added as a positive control for full-length FFV IN DNA (1,149bp); Δstu-Ι FIN, Δstu-Ι FIN only added as a positive control for amplification of deleted FFV IN DNA (864bp); MOCK, chromosomal DNA extracted from uninfected CRFK cells; DNA extract, extracted chromosomal DNA added as a target template at 100 ng; DNA extract +Δstu-Ι FIN and Δstu-Ι FIN 10−2–10−9, Δstu-Ι FIN DNA added at 10-fold serial dilutions for competitive PCR reaction. (b) Failure to detect viral DNA in the chromosomal DNA of the D107A, D164E, and E200D virus-infected cells. CRFK cells were infected with the 3 days post-transfection culture supernatant of D107A, D164E, and E200D, with volume of the culture supernatants used for infection being equal to that of WT infection. At 24 and 48 h post-infection, chromosomal DNAs were prepared as mentioned above. To detect viral DNA integrated into chromosomal DNA, PCR amplification was performed using the extracted chromosomal DNA of various amounts (100–1000 ng) as the template DNA.
Discussion
We have reported the in vitro analysis of nine FFV IN mutants containing single amino acid substitution at a highly conserved residue and close to the invariant residue positions in the CCD of IN. In addition to the highly conserved DDE residues, we selected adjacent amino acid residues from the DD(35)E motif region for determining their potentially significant role for activities of IN. D107, D164, and E200 residue positions were selected because of their conservation and homology to the HIV. Q165, Y191, and S195 were selected because they correspond to N117, Y143, and S147 of HIV-1 IN, respectively, which are antiviral-drug resistant variants, particularly for IN strand transfer inhibitors. IN is active as a multimer in vivo as well as in vitro with two divalent metal ions [Citation38,Citation39] and DD(35)E motif is highly involved in this situation. Therefore it was assumed that IN enzymatic activity may be slightly changed without complete loss if one of DD(35)E residue is replaced by another acidic amino acid residue, because highly conserved region is acidic triad as ever. Therefore, we made 2 single mutation for each DDE residue position, one is alanine and the other is similar acidic amino acid.
Generally, in vitro studies of the enzymatic activities of purified retroviral IN have provided the following model of the role of the IN protein in retroviral integration reaction. In agreement with other retroviral DDE motif research data [Citation40–Citation42], our results displayed a critical role of D107, D164, and E200 residue positions in the catalytic functions of IN. Amino acid substitution of these residues could affect most activities of IN due to several possible mechanisms, including conformational changes in the FFV IN complex that affects steric hindrance, loss of stabilizing binding interactions, and divalent cation binding [Citation43,Citation44]. So we expected a difference in the catalytic activities between two types of DDE mutations, but there were no significant differences of enzymatic activities.
In the case of Q165A, Y191A, and S195A, they showed IN catalytic activities with somewhat higher level in strand transfer and disintegration in vitro assay. These three residues are not highly conserved, but they can be influential in IN reaction and viral replication because they are located closely to DD(35)E highly conserved region that is important for IN multimerization and metal ion binding.
The results of transfection experiments using COS-1 cells suggest that single residue mutation in any amino acid residue in CCD exhibits no crucial effect on viral particle formation. However, based on infection experiments using CRFK cells, the mutants D107A, D107E, D164A, D164E, E200A, and E200D showed a significant reduction in infectivity, whereas the virions containing the IN mutation Q165A, Y191A, and S195A replicated normally, although the rate was less than that of WT. Impairment of the DD(35)E motif of FFV IN appeared to render virions harboring mutant INs unable to replicate normally in the cellular culture. Taken together, our data clearly support the fundamental role of the DD(35)E motif in the catalytic functions of IN and suggest that it is essential for the infectivity of FFV virions. Thus, it becomes very interesting to further study D107, D164, and E200 residues and other positions close to the DD(35)E conserved region for a better understanding of their exact role, particularly viral particle formation and infectivity.
We used competitive PCR assay for quantification of integrated viral load present in the infected CRFK cells. Competitive PCR method can discriminate integration frequency between WT and mutants quick and easily. Provirus harboring Q165A, Y191A, and S195A FFV IN mutation was detected at reduced amounts, 10–100-fold less efficiently in 24 h post-infection and 100–1000-fold less efficiently in 48 h post-infection, compared to that of WT. In agreement with our previous data, D107A, D164E, and E200D FFV IN mutant provirus were not detected because non-integrated DNA may have been already degraded or diluted via mitosis. Therefore, the data clearly suggest that amino acid substitution in CCD, particularly the DD(35)E motif, result in non-infectious virions, preventing the integration of the viral DNA into the host chromosome of infected cells and precluding the formation of new infectious virions.
Conclusions
We report the first case of single residue mutation of the DDE motif into either alanine or similar acidic amino acid residues in FFV IN. We showed that impairment of the DDE motif profoundly affected FFV IN enzymatic activities. In addition, other mutations such as Q165A, Y191A, and S195A also reduced catalytic effects. As expected, the DDE motif was observed to be critical for polynucleotidyl transfer reactions, including 3′-end processing, strand transfer and disintegration. Therefore, we could not detect virus titer and production with DDE mutant viruses, because the DDE mutant viruses cannot integrate their cDNA into host chromosome. This study, together with previous reports of FFV IN CTD [Citation45], demonstrated that the FFV IN DDE motif is essential for efficient viral replication like other retrovirus IN proteins. In the case of Q165A, Y191A, and S195A, all three single point mutation exhibited declined IN activities at different extents. These three amino acid residues are not the primary active sites of FFV IN, but their substitutions had a negative effect on IN catalytic activities.
With increasing use of foamy viral vectors and availability for antiviral drug model virus, other mutations in FFV IN should be studied to further increase our understanding on FFV IN enzymatic activities and how they affect feline foamy viral replication cycle. This study offers an insight into the similar structural and functional relations within mammalian foamy viral IN, including human, bovine, simian, and feline foamy viral IN.
Author contribution
GE Lee performed most of the research works, designed and carried out experiments, and wrote the manuscript. J Kim helped DNA construction and virus measurements. CG Shin supervised the overall research and wrote the manuscript.
Table_S1.docx
Download MS Word (20.4 KB)Supplemenatry_material_captions-h.docx
Download MS Word (253.4 KB)Acknowledgments
This work was supported by a grant from the National Research Foundation of Korea (NRF) funded by the Korean government (NRF-2015R1D1A1A01059592) to Cha-Gyun Shin. The molecular clone of feline foamy virus (pCF-7) and FeFAB cell for FeFAB assay were kind gifts from Dr. Martin Lochelt, German Cancer Research Center, Germany.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Coffin J, Hughes S, Varmus H. Retroviruses. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997.
- Francki R, Fauquet C, Knudson D, et al. Classification and nomenclature of viruses: fifth report of the international committee on taxonomy of viruses. Virology division of the international union of microbiological societies. Vol. 2, Berlin, Germany: Springer Science & Business Media; 2012.
- Lindemann D, Rethwilm A. Foamy virus biology and its application for vector development. Viruses. 2011;3(5):561–585.
- Rethwilm A. Foamy virus vectors: an awaited alternative to gammaretro-and lentiviral vectors. Curr Gene Ther. 2007;7(4):261–271.
- Trobridge GD. Foamy virus vectors for gene transfer. Expert Opin Biol Ther. 2009;9(11):1427–1436.
- Hare S, Gupta SS, Valkov E, et al Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464(7286):232.
- Maertens GN, Hare S, Cherepanov P. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature. 2010;468(7321):326.
- Asante-Appiah E, Skalka AM. Molecular mechanisms in retrovirus DNA integration. Antiviral Res. 1997;36(3):139–156.
- Mizuuchi K. Polynucleotidyl transfer reactions in transpositional DNA recombination. J Biol Chem. 1992;267:21273–21276.
- Brown PO, Bowerman B, Varmus HE, et al Retroviral integration: structure of the initial covalent product and its precursor, and a role for the viral IN protein. Proc Natl Acad Sci. 1989;86(8):2525–2529.
- Fujiwara T, Mizuuchi K. Retroviral DNA integration: structure of an integration intermediate. Cell. 1988;54(4):497–504.
- Engelman A, Mizuuchi K, Craigie R. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell. 1991;67(6):1211–1221.
- Gerton JL, Herschlag D, Brown PO. Stereospecificity of reactions catalyzed by HIV-1 integrase. J Biol Chem. 1999;274(47):33480–33487.
- Daniel R, Katz RA, Skalka AM. A role for DNA-PK in retroviral DNA integration. Science. 1999;284(5414):644–647.
- Drelich M, Wilhelm R, Mous J. Identification of amino acid residues critical for endonuclease and integration activities of HIV-1 IN protein in vitro. Virology. 1992;188(2):459–468.
- Bujacz G, Jaskólski M, Alexandratos J, et al The catalytic domain of avian sarcoma virus integrase: conformation of the active-site residues in the presence of divalent cations. Structure. 1996;4(1):89–96.
- Leh H, Brodin P, Bischerour J, et al Determinants of Mg2+-dependent activities of recombinant human immunodeficiency virus type 1 integrase. Biochemistry. 2000;39(31):9285–9294.
- Bouyac-Bertoia M, Dvorin JD, Fouchier RA, et al HIV-1 infection requires a functional integrase NLS. Mol Cell. 2001;7(5):1025–1035.
- Engelman A, Englund G, Orenstein JM, et al Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J Virol. 1995;69(5):2729–2736.
- Shin C-G, Taddeo B, Haseltine WA, et al Genetic analysis of the human immunodeficiency virus type 1 integrase protein. J Virol. 1994;68(3):1633–1642.
- Valkov E, Gupta SS, Hare S, et al Functional and structural characterization of the integrase from the prototype foamy virus. Nucleic Acids Res. 2008;37(1):243–255.
- Enssle J, Moebes A, Heinkelein M, et al An active foamy virus integrase is required for virus replication. J Gen Virol. 1999;80(6):1445–1452.
- Pinto-Santini DM, Stenbak CR, Linial ML. Foamy virus zoonotic infections. Retrovirology. 2017;14(1):55.
- Switzer WM, Bhullar V, Shanmugam V, et al Frequent simian foamy virus infection in persons occupationally exposed to nonhuman primates. J Virol. 2004;78(6):2780–2789.
- Schwantes A, Ortlepp I, Löchelt M. Construction and functional characterization of feline foamy virus-based retroviral vectors. Virology. 2002;301(1):53–63.
- Löchelt M, Muranyi W, Flügel R. Human foamy virus genome possesses an internal, Bel-1-dependent and functional promoter. Proc Natl Acad Sci. 1993;90(15):7317–7321.
- Horton RM, Ho SN, Pullen JK, et al. Gene splicing by overlap extension. Recombinant DNA methodology II. MA, USA: Elsevier; 1995. p. 363–372.
- Gerton JL, Ohgi S, Olsen M, et al Effects of mutations in residues near the active site of human immunodeficiency virus type 1 integrase on specific enzyme-substrate interactions. J Virol. 1998;72(6):5046–5055.
- Métifiot M, Vandegraaff N, Maddali K, et al Elvitegravir overcomes resistance to raltegravir induced by integrase mutation Y143. Aids. 2011;25(9):1175–1178.
- Shimura K, Kodama E, Sakagami Y, et al Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J Virol. 2008;82(2):764–774.
- Chaga G, Hopp J, Nelson P. Immobilized metal ion affinity chromatography on Co2+‐carboxymethylaspartate–agarose Superflow, as demonstrated by one‐step purification of lactate dehydrogenase from chicken breast muscle. Biotechnol Appl Biochem. 1999;29(1):19–24.
- Engelman A, Cherepanov P. Retroviral integrase structure and DNA recombination mechanism. Microbiol Spectrum. 2014;2(6):1.
- Enssle J, Fischer N, Moebes A, et al Carboxy-terminal cleavage of the human foamy virus Gag precursor molecule is an essential step in the viral life cycle. J Virol. 1997;71(10):7312–7317.
- Hamann MV, Müllers E, Reh J, et al The cooperative function of arginine residues in the Prototype Foamy Virus Gag C-terminus mediates viral and cellular RNA encapsidation. Retrovirology. 2014;11(1):87.
- Spannaus R, Schneider A, Hartl MJ, et al Foamy virus Gag p71-p68 cleavage is required for template switch of the reverse transcriptase. J Virol. 2013;87(13):7774–7776.
- Hamid FB, Kim J, Shin C-G. Cellular and viral determinants of retroviral nuclear entry. Can J Microbiol. 2015;62(1):1–15.
- Hamid FB, Kim J, Shin C-G. Distribution and fate of HIV-1 unintegrated DNA species: a comprehensive update. AIDS Res Ther. 2017;14(1):9.
- Jones K, Coleman J, Merkel G, et al Retroviral integrase functions as a multimer and can turn over catalytically. J Biol Chem. 1992;267(23):16037–16040.
- Fletcher TM, Soares MA, McPhearson S, et al Complementation of integrase function in HIV-1 virions. Embo J. 1997;16(16):5123–5138.
- Kulkosky J, Jones KS, Katz RA, et al Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol Cell Biol. 1992;12(5):2331–2338.
- Shibagaki Y, Holmes ML, Appa RS, et al Characterization of feline immunodeficiency virus integrase and analysis of functional domains. Virology. 1997;230(1):1–10.
- Bujacz G, Alexandratos J, Wlodawer A, et al Binding of different divalent cations to the active site of avian sarcoma virus integrase and their effects on enzymatic activity. J Biol Chem. 1997;272(29):18161–18168.
- Grobler JA, Stillmock K, Hu B, et al Diketo acid inhibitor mechanism and HIV-1 integrase: implications for metal binding in the active site of phosphotransferase enzymes. Proc Natl Acad Sci. 2002;99(10):6661–6666.
- Wang JY, Ling H, Yang W, et al Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. Embo J. 2001;20(24):7333–7343.
- Kim J, Lee G-E, Lochelt M, et al Integrase C-terminal residues determine the efficiency of feline foamy viral DNA integration. Virology. 2018;514:50–56.