
ABSTRACT
Fatty acid monoesters of hydroxytyrosol [2-(3,4-dihydroxyphenyl)ethanol] were synthesized in two steps from tyrosol (4-hydroxyphenylethanol) by successive Candida antarctica lipase B-catalyzed chemoselective acylation on the primary aliphatic hydroxy group over phenolic hydroxy group in tyrosol, and 2-iodoxybenzoic acid (IBX)-mediated hydroxylation adjacent to the remaining free phenolic hydroxy group. Examination of their suppression effects on nitric oxide production stimulated by lipopolysaccharides in RAW264.7 cells showed that hydroxytyrosol butyrate exhibited the highest inhibition (IC50 7.0 μM) among the tested compounds.
Graphical Abstract
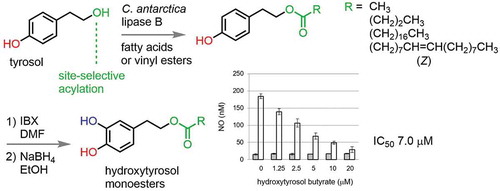
Hydroxytyrosol monoesters were site-selectively synthesized from tyrosol in two steps. Among them, butyrate showed strongest suppression effect on NO production stimulated by LPS.
Oleuropein (1a) and its hydrolysate, hydroxytyrosol (1b, ) are representative antioxidative catechols in olives [Citation1]. A variety of physiological activities of 1b have been reported [Citation2–Citation15]. For example, 1b exhibited anti-inflammatory activity by preventing nitric oxide (NO) production stimulated by lipopolysaccharides (LPS) in macrophages [Citation15]. Recently, one of the authors (M. F.-T.) has investigated the protective effect of 1b and its ester derivatives 1c and 1d against neuronal cells apoptosis induced by the Parkinson’s disease-related neurotoxin 6-hydroxydopamine (6-OHDA) in SH-SY5 cells, with butyrate 1d showing the strongest inhibition. Only 1d among three compounds induced the expression of the transcription factor, NF-E2-related factor-2 (Nrf2) and enhanced its transcriptional activation in those cells [Citation16]. Independently, they found that Nrf2 suppressed the LPS-induced transcriptional up-regulation of the proinflammatory cytokines and the inducible NO synthase (iNOS) [Citation17]. We became interested in the inhibitory effect of esters 1c–f in NO generation in the murine macrophage RAW 264.7 cell line. For longer chain fatty acid esters 1e (stearate) and 1f (oleate), which are known as antioxidants [Citation18–Citation22], the esterification with long chain fatty acids was expected to increase the effectiveness [Citation23,Citation24].
Scheme 1. Catechols involved in olives 1a and 1b, examples of fatty acid monoesters 1c to 1f derived from 1a, and the previous synthesis of 1d.
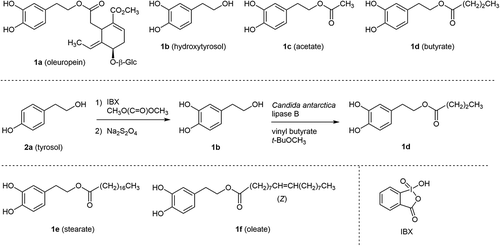
For the biological assay, the efficient preparation of fatty acid esters 1c–f was demanded. In the previous study [Citation16], butyrate 1d was synthesized by lipase-mediated chemoselective acylation of primary aliphatic hydroxy group over phenolic hydroxy groups in commercially available 1b [Citation25,Citation26] as shown in .
However, 1b itself is an expensive starting material, although it can be supplied either by the extraction and purification of wastewater from the manufacture of olive oil [Citation12,Citation27–Citation30]. Chemists’ efforts have been devoted to the synthesis of 1b [Citation30–Citation33]. Among them, the site-selective hydroxylation of tyrosol (2a) with 2-iodoxybenzoic acid (IBX) as shown in is straightforward. The reported yield, however, was only 30% due to the difficulty in isolation of the desired product 1b. Those authors insisted that either increased lipophilicity of substrates or use of the elaborated polymer-supported IBX derivative [Citation33] is necessary, to acquire an enhanced yield.
To solve above-mentioned problems, we herein describe alternative routes for the synthesis of 1c–f from readily available 2a, by exchanging the order of hydroxylation and acylation.
Results and discussion
Candida antarctica lipase B-catalyzed acylation of 2a has so far been reported [Citation22,Citation34]. In our case, the desired reaction proceeded smoothly by employing a short chain fatty acid vinyl ester to give the corresponding acetate 2b and butyrate 2c in a quantitative manner. Hydroxylation adjacent to the phenolic hydroxy group was performed by IBX-mediated oxidation [Citation21] and subsequent reduction with sodium borohydride [Citation35]. The desired products 1c (at 88% conversion) and 1d (at 91% conversion) were isolated in 62% and 58% yields, respectively ().
Scheme 2. Chemoenzymatic synthesis of 1c - f starting from readily available tyrosol 2a.
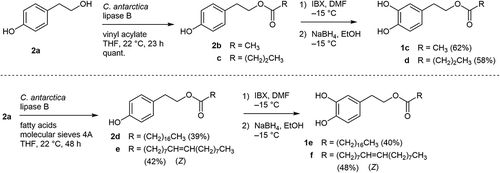
Due to limited availability of long chain fatty acid vinyl esters with high purity, we adopted free fatty acids for the lipase-catalyzed acylation [Citation25] in the synthesis of stearate 2d and oleate 2e. Molecular sieves 4A were added to remove water that was concomitantly formed during the esterification. The reactions were slow even under such forced conditions and required prolonged reaction times (48 h) at room temperature, compared with the reactions of the short chain vinyl esters. At a conversion of around 50%, the reaction was worked up, and the desired products were purified by medium-pressure liquid chromatography. Monoesters 2c and 2d were isolated in 39% and 42% yields, respectively (). Although the hydroxylation yields were somewhat lower, stearate 1e and oleate 1f were isolated in 40% and 48% yields in sufficient amounts for the subsequent evaluation of inhibitory effects on NO production stimulated by LPS in RAW264.7 cells.
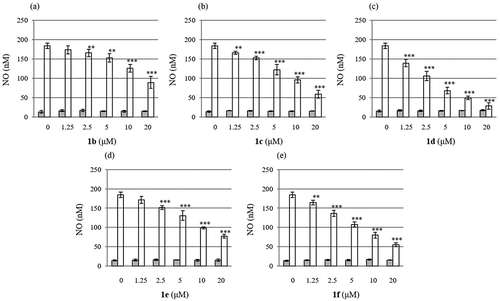
The results for the suppression effects of 1b-f on NO production are summarized in . Judging from the bar charts, butyrate 1d exhibited the strongest inhibitory effect ()) among the tested compounds. The estimated IC50 values (μM) for each compound were 18.5 for 1b, 12.7 for 1c, 7.0 for 1d, 14.5 for 1e, and 11.2 for 1f, respectively. It should be noted that none of the compounds used in this study caused cell death, indicating that their inhibitory effects on NO production were not due to their cytotoxicity.
Figure 1. Suppression effect of 1b - f on the NO production stimulated by LPS in RAW264.7 cells. Shaded (left) bars and open (right) bars represent concentration of NO without (control experiments) or with LPS, respectively. Values are given as the mean ± S.D. from four independent experiments. Multiple group comparisons were made using one- or two-way analysis of variance (ANOVA) followed by Tukey’s test. *P < 0.05; **P < 0.01; ***P < 0.001 significantly different from the control group stimulated with LPS. For detail, see experimental section.
We state here only a brief comment for the proposed mechanism of the inhibition. As mentioned in the introduction [Citation16,Citation17], butyrate 1d induced the expression of Nrf2 and enhanced its transcriptional activation in the cells. Also, Nrf2 interfered with the LPS-induced transcriptional up-regulation of the proinflammatory cytokines and inducible NO synthase (iNOS) in RAW264.7 cells. From these findings, we suppose that butyrate 1d effectively inhibited NO production by activating Nrf2.
The action of intracellular short-chain fatty acid esterases on 1b–d would promptly converge them into the identical free form 1b. The difference in inhibitory effect among those of compounds is probably ascribable to the cell permeability depending upon the lipophilicity of compounds. The synthesis and evaluation of acylated forms with fatty acids of intermediate chain length between 1d and 1e may provide a clue to clarify this phenomenon.
Conclusion
We synthesized four fatty acid monoesters (1c-f) of hydroxytyrosol (1b) from inexpensive tyrosol (2a) by combining lipase-catalyzed chemoselective acylation and subsequent hydroxylation. By arranging the order of these two reactions in this way, the total yield of the products and the handling of the intermediates were improved from those in the originally reported procedure [Citation25,Citation26]. All products showed inhibitory effects on NO production stimulated by LPS. Among them, butyrate 1d was the most potent inhibitor with IC50 value of 7.0 μM. In this way, we found a new property of hydroxytyrosol monoesters other than so-far reported anti-apoptosis effect.
Experimental
General
Candida antarctica lipase B (Novozym 435) was purchased from Novozymes Japan. 2-(4-Hydroxyphenyl)ethanol (tyrosol, H0720) was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). RAW264.7 cells were purchased from the Riken Cell Bank (Ibaraki, Japan). LPS (E. coli 055:B5) was purchased from Sigma-Aldrich Co. (St, Louis, MO, USA). Dulbecco’s Modified Eagle’s Medium (DMEM) and a penicillin (10,000 units/mL)-Streptomycin (1 mg/mL) mixed solution were purchased from Nacalai Tesque Inc. (Kyoto, Japan). Fetal bovine serum (FBS, Gibco®) was purchased from Thermo Fisher Scientific Inc. (Waltham MA, USA). (Nacalai Tesque). Column chromatography was performed with silica gel (Kanto Chemical Co. Silica Gel 60 N 37560-79, spherical and neutral, 40–50 µm). Medium-pressure liquid chromatography was performed using Silica Gel 60 (spherical and neutral; 100–210 µm, 37560-79, Kanto Chemical Co.) and the Isolera One flash purification system (Biotage, Sweden). Preparative TLC was performed with Merck Silica Gel 60 F254 plates (0.5 mm thickness, No. 1.05744.0001). 1H NMR spectra were measured at 400 MHz on a VARIAN 400-MR or at 500 MHz on an Agilent INOVA-500 spectrometer and 13C NMR spectra were measured at 100 MHz on a VARIAN 400-MR or at 125 MHz on an Agilent INOVA-500 spectrometer. DMSO-d6 and CDCl3 were used as a solvent and the residual peaks were used as an internal standard (1H NMR: DMSO-d6 2.48 ppm, CHCl3 in CDCl3 7.26 ppm; 13C NMR: DMSO-d6 39.9 ppm, CDCl3 77.0 ppm). IR spectra were measured as ATR on a Jasco FT/IR-4700 FT-IR spectrometer. High resolution mass spectra (HRMS) were measured by a on Jeol JMS-T100LP AccuTOF. Microplate reader Infinite M1000 (Tecan Group Ltd. Tokyo Japan) was used for the measurement of absorbance.
2-(4-Hydroxylphenyl)ethyl acetate (2b)
To a solution of 2a (2.8 g, 20 mmol) in tetrahydrofuran (THF, 37 mL) was added C. antarctica lipase B (409 mg) and vinyl acetate (2.8 mL, 30 mmol). The mixture was stirred for 4 h at room temperature. The mixture was diluted with chloroform and filtered with a pad of Celite. The combined filtrate and washings were dried in vacuo to give 2b as colorless solid (3.84 g, quantitative). M.p. 59–60°C. 1H-NMR (400 MHz, DMSO- d6) δ: 1.96 (3H, s), 2.73 (2H, t, J = 7.0 Hz), 4.10 (2H, t, J = 7.0 Hz), 6.66 (2H, d, J = 8.4 Hz), 7.00 (2H, d, J = 8.4 Hz), 9.19 (s, OH). 13C-NMR (100 MHz, DMSO-d6) δ: 21.2, 34.0, 65.2, 115.6, 128.3, 130.2, 156.3, 170.8. IR νmax cm–1: 3361, 3022, 2957, 1705, 1614, 1596, 1515, 1444, 1388, 1364, 1227, 1173, 1107, 1030, 975, 830. HR-MS [ESI+, (M+Na)+] calculated for C10H12O3Na, 203.0684; found, 203.0707.
2-(4-Hydroxylphenyl)ethyl butyrate (2c)
In a similar manner as described for the synthesis of 2b in the previous section, diol 2a was treated with C. antarctica lipase B and vinyl butyrate. The workup and purification furnished 2c as pale yellow oil (5.36 g, quantitative). 1H-NMR (400 MHz, DMSO-d6) δ: 0.84 (3H, t, J = 7.4 Hz), 1.51 (2H, dq, J = 7.3, 7.4 Hz), 2.23 (2H, t, J = 7.3 Hz), 2.75 (2H, t, J = 7.0 Hz), 4.14 (2H, t, J = 7.0 Hz), 6.67 (2H, d, J = 8.6 Hz), 7.02 (2H, d, J = 8.6 Hz), 9.21 (OH, s). 13C-NMR (125 MHz, DMSO-d6) δ: 13.8, 18.4, 34.0, 35.8, 65.0, 115.6, 128.3, 130.2, 156.3, 173.1. IR νmax cm−1 3380, 2963, 2935, 2875, 1704, 1614, 1596, 1515, 1445, 1389, 1356, 1308, 1185, 1171, 1105, 1046, 985, 828. HR-MS [ESI+, (M+Na)+]: calculated for C12H16O3Na 231.0997; found, 231.1001.
2-(4-Hydroxylphenyl)ethyl stearate (2d)
To a solution of 2a (150 mg, 1.1 mmol) in THF (1.5 mL) was added C. antarctica lipase B (15 mg), molecular sieves 4A (50 mg), and stearic acid (460 mg, 1.6 mmol). The mixture was stirred for 48 h at room temperature. The mixture was diluted with chloroform and filtered with a pad of Celite. The combined filtrate and washings were dried in vacuo. The residue was purified by medium-pressure liquid chromatography. Elution with hexane-ethyl acetate (10:1) furnished 2d as colorless amorphous solid (172 mg, 39%). 1H-NMR (500 MHz, CDCl3) δ: 0.88 (3H, t, J = 7.0 Hz), 1.16–1.34 (28H, m), 1.51–1.65 (2H, m), 2.28 (2H, t, J = 7.6 Hz), 2.86 (2H, t, J = 7.1 Hz), 4.24 (2H, t, J = 7.1 Hz), 6.66 (2H, d, J = 8.4 Hz), 7.00 (2H, d, J = 8.4 Hz). Due to the broadening of signal, proton on phenolic hydroxy group was not detected. 13C-NMR (125 MHz, CDCl3) δ: 14.1, 22.7, 24.9, 29.1, 29.3, 29.4, 29.5, 29.6, 29.7, 29.7, 31.9, 34.3, 34.4, 65.0, 115.3, 130.0, 154.2, 174.0. Some signals of methylene carbons were overlapped. IR νmax cm–1: 3252, 2915, 2847, 2359, 1731, 1615, 1519, 1464, 1392, 1251, 1234, 1214, 1193, 1163, 826, 719, 564, 521. HR-MS [ESI+, (M+Na)+] calculated for C26H44O3Na, 427.3188; found, 427.3171.
2-(4-Hydroxylphenyl)ethyl oleate (2e)
In a similar manner as described for the synthesis of 2d in the previous section, diol 2a was treated with C. antarctica lipase B and oleic acid. The workup and purification furnished 2e as colorless amorphous solid (180 mg, 42%). 1H-NMR (500 MHz, CDCl3) δ: 0.81 (3H, t, J = 6.9 Hz), 1.16–1.34 (20H), 1.43–1.63 (2H, m), 1.87–2.01 (4H, m), 2.21 (2H, t, J = 7.5 Hz), 2.78 (2H, t, J = 7.1 Hz), 4.17 (2H, t, J = 7.1 Hz), 5.10–5.44 (2H, m), 6.68 (2H, d, J = 8.4 Hz), 7.00 (2H, d, J = 8.4 Hz). Due to the broadening of signal, proton on phenolic hydroxy group was not detected. 13C-NMR (125 MHz, CDCl3) δ: 14.1, 22.7, 24.9, 27.2, 27.2, 29.1, 29.2, 29.3, 29.5, 29.7, 29.8, 31.9, 34.3, 34.4, 65.0, 115.3, 129.8, 130.0, 130.0, 154.3, 174.0. Some signals of methylene carbons were overlapped. IR νmax cm–1: 3389, 3004, 2923, 2852, 2359, 2342, 1737, 1710, 1615, 1596, 1517, 1456, 1354, 1225, 1172, 1006, 830, 724. HR-MS [ESI+, (M+Na)+] calculated for C26H42O3Na, 425.3032; found, 425.3035.
2-(3,4-Dihydroxylphenyl)ethyl acetate (1c)
IBX (3.34 g, 45% purity, 5.4 mmol) was sonicated in N,N-dimethylformamide (DMF, 128 mL). The resulting suspension was stirred at – 15°C under Ar, and to that was added a solution of 2b (724 mg, 4.0 mmol) in DMF (12 mL) in one portion. The mixture was further stirred for 22 h at – 15°C under Ar. To the mixture, a pre-cooled solution of sodium borohydride (NaBH4, 120 mg) in ethanol (8.0 mL) was slowly added at – 15°C, and the color of the mixture turned from brown to yellow. The mixture was diluted with ethyl acetate (180 mL) and to that was added a solution of 0.1 M phosphate buffer with 3.5% of sodium dithionite and 35% of sodium chloride. The mixture was stirred at room temperature for 1.5 h. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate twice. The combined extract was washed with aqueous solution including sodium hydrogen carbonate (10%) and sodium dithionite (5%) and brine, dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by silica gel column chromatography. Elution with hexane-ethyl acetate (5:1 to 3:1) furnished 1c as a pale yellow oil (489 mg, conversion: 85%; yield: 62%). 1H-NMR (500 MHz, DMSO-d6) δ: 1.96 (3H, s), 2.67 (2H, t, J = 7.1 Hz), 4.08 (2H, t, J = 7.1 Hz), 6.45 (1H, dd, J = 2.2, 8.0 Hz), 6.59 (1H, d, J = 2.2 Hz), 6.62 (1H, d, J = 8.0 Hz), 8.67 (OH, s), 8.75 (OH, s). 13C-NMR (100 MHz, DMSO-d6) δ: 21.2, 34.2, 65.2, 116.0, 116.6, 119.9, 129.0, 144.2, 145.5, 170.8. IR νmax cm−1 3360, 3035, 2959, 1703, 1605, 1519, 1444, 1387, 1363, 1241, 1192, 1149, 1113, 1031, 978, 956, 864, 807, 782. HR-MS [ESI+, (M+Na)+]: calculated for C10H12O4Na, 219.0633; found, 219.0671.
2-(3,4-Dihydroxylphenyl)ethyl butyrate (1d)
In a similar manner as described for the synthesis of 1c in the previous section, butyrate 2c (418 mg, 2.0 mmol) was treated with IBX then NaBH4. The workup and purification furnished 1d as colorless oil (261 mg, conversion: 91%; yield: 58%). 1H-NMR (400 MHz, DMSO-d6) δ: 0.85 (3H, t, J = 7.3 Hz), 1.51 (2H, tq, J = 7.3, 7.3 Hz), 2.24 (2H, t, J = 7.3 Hz), 2.69 (2H, t, J = 7.1 Hz), 4.12 (2H, t, J = 7.1 Hz), 6.46 (1H, dd, J = 2.2, 7.8 Hz), 6.60 (1H, d, J = 2.2 Hz), 6.63 (1H, d, J = 7.8 Hz), 8.69 (OH, s), 8.76 (OH, s). 13C-NMR (125 MHz, DMSO-d6) δ: 13.8, 18.4, 34.3, 35.8, 65.0, 115.9, 116.6, 119.9, 129.0, 144.2, 145.5, 173.2. IR νmax cm−1 3378, 3035, 2963, 2934, 2875, 1703, 1605, 1520, 1444, 1384, 1346, 1279, 1256, 1184, 1112, 1044, 986, 956, 923, 865, 807, 782. HR-MS [ESI+, (M+Na)+]: calculated for C12H16O4Na, 247.0946; found, 247.0938.
2-(3,4-Dihydroxylphenyl)ethyl stearate (1e)
In a similar manner as described for the synthesis of 1c in the previous section, stearate 2d (221 mg, 0.55 mmol) was treated with IBX then NaBH4. The workup and purification furnished 1e as colorless amorphous solid (92 mg, yield: 40%). 1H-NMR (500 MHz, CDCl3) δ: 0.81 (3H, t, J = 7.0 Hz), 1.07–1.32 (28H, m), 1.51–1.65 (2H, m), 2.22 (2H, t, J = 7.6 Hz), 2.75 (2H, t, J = 7.1 Hz), 4.16 (2H, t, J = 7.1 Hz), 5.13 (1H, broad), 5.30 (1H, broad), 6.57 (1H, dd, J = 2.0, 8.0 Hz), 6.67 (1H, d, J = 2.0 Hz), 6.72 (1H, d, J = 8.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 14.1, 22.7, 24.9, 29.1, 29.3, 29.4, 29.7, 29.7, 31.9, 34.4, 34.4, 64.8, 115.3, 116.0, 121.4, 130.9, 142.1, 143.5, 174.0. Some signals of methylene carbons were overlapped. IR νmax cm–1: 3321, 2954, 2915, 2848, 1737, 1598, 1520, 1462, 1330, 1275, 1254, 1234, 1213, 1192, 1173, 1119, 961, 822, 728. HR-MS [ESI+, (M+Na)+] calculated for C26H44O4Na, 443.3137; found, 443.3159.
2-(3,4-Dihydroxylphenyl)ethyl oleate (1f)
In a similar manner as described for the synthesis of 1c in the previous section, oleate 2f (120 mg, 0.3 mmol) was treated with IBX then NaBH4. The workup and purification furnished 1f as a colorless oil (60 mg, yield: 48%). 1H-NMR (500 MHz, CDCl3) δ: 0.88 (3H, t, J = 7.0 Hz), 1.07–1.32 (20H, m), 1.51–1.63 (2H, m), 1.92–2.10 (4H, m), 2.29 (2H, t, J = 7.6 Hz), 2.81 (2H, t, J = 7.1 Hz), 4.24 (2H, t, J = 7.1 Hz), 5.23–5.45 (2H, m), 6.64 (1H, dd, J = 1.5, 8.0 Hz), 6.73 (1H, d, J = 1.5 Hz), 6.78 (1H, d, J = 8.0 Hz). Due to the broadening of signal, proton on phenolic hydroxy group was not detected. 13C-NMR (125 MHz, CDCl3) δ: 14.1, 22.7, 24.9, 27.2, 29.1, 29.2, 29.3, 29.5, 29.7, 29.8, 31.9, 34.4, 34.4, 64.9, 115.3, 115.9, 121.3, 129.8, 130.0, 130.8, 142.1, 143.5, 174.1. Some signals of methylene carbons were overlapped. IR νmax cm–1: 3334, 3004, 2918, 2849, 2360, 2341, 1732, 1706, 1604, 1520, 1463, 1276, 1252, 1214, 1179, 1113, 813, 720, 504. HR-MS [ESI+, (M+Na)+] calculated for C26H44O4Na, 441.2981; found, 441.2990.
Examination of the effect of 1b–1f on the production of NO mediated by LPS
The murine macrophage cell line, RAW264.7 was cultured at 37°C under 5% CO2/95% air in DMEM supplemented with 10% FBS and penicillin-streptomycin mixed solution. RAW264.7 cells (2 × 105 cells) were cultured in a 24-well plate and pre-incubated with various concentrations of hydroxytyrosol and its derivatives (1b-f) or a solution of dimethyl sulfoxide (0.1%) at 37°C for 1 h prior to the stimulation with LPS (1 μg/mL) for 24 h. The concentration of nitrirte (NO2–) ion derived from NO in the culture supernatants was estimated with microplate reader at the absorbance at 540 nm, after coloration using Griess reagent, which contained sulfanilamide (1%), N-naphthylethylenediamine (0.1%), and H3PO4 (2.5%). IC50 values were calculated from the concentration of 1b-f which was required for 50% inhibition of NO production in the experiments. The results were expressed as the bar charts in .
Author contributions
T.S and M.F-T. designed this study; A.S. and M.P. mainly carried out the experiments in chemistry; A.N. mainly carried out the experiment in biology; T.S. and M.F.-T. wrote the manuscript with assistance from all authors; and K.H., S.H. and H.T. occasionally discussed on the results.
Acknowledgments
M.P. thanks both of Graduate School of SIGMA Clermont and Graduate School of Pharmaceutical Sciences, Keio University, to participate in this study as a visiting research student under agreement from May to September in 2017.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Visioli F, Galli C. Olive oil phenols and their potential effects on human health. J Agric Food Chem. 1998;46:4292–4296.
- Salami M, Galli C, De Angelis L, et al Formation of F2-isoprostanes in oxidized low density lipoprotein: inhibitory effect of hydroxytyrosol. Phamacol Res. 1995;31:275–279.
- Petroni A, Blasevich M, Salami M, et al Inhibition of platelet aggregation and eicosanoid production by phenolic components of olive oil. Thromb Res. 1995;78:151–160.
- Manna C, Galletti P, Cucciolla V, et al The protective effect of the olive oil polyphenol (3,4-dihydroxyphenyl)ethanol counteracts reactive oxygen metabolite-induced cytotoxicity in Caco-2 cells. J Nutr. 1997;127:286–292.
- Kohyama N, Nagata T, Fujimoto S, et al Inhibition of arachidonate lipoxygenase activities by 2-(3,4-dihydroxyphenyl)ethanol, a phenolic compound from olives. Biosci Biotechnol Biochem. 1997;61:347–350.
- Aruoma OI, Deiana M, Jenner A, et al Effect of hydroxytyrosol found in extra virgin olive oil on oxidative DNA damage and on low-density lipoprotein oxidation. J Agric Food Chem. 1998;46:5181–5187.
- Manna C, Galletti P, Cucciolla V, et al Olive oil hydroxytyrosol protects human erythrocytes against oxidative damages. J Nutr Biochem. 1999;10:159–165.
- Visioli F, Galli C, Plasmati E, et al Olive phenol hydroxytyrosol prevents passive smoking-induced oxidative stress. Circulation. 2000;102:2169–2171.
- Ragione FD, Cucciolla V, Borriello A, et al Hydroxytyrosol, a natural molecule occurring in olive oil, induces cytochrome c-dependent apoptosis. Biochem Biophys Res Commun. 2000;278:733–739.
- Fabiani R, De Bartolomeo A, Rosignoli P, et al Cancer chemoprevention by hydroxytyrosol isolated from virgin olive oil through G1 cell cycle arrest and apoptosis. Eur J Cancer Prev. 2002;11:351–358.
- Palmerini CA, Carlini E, Saccardi C, et al Activity of olive oil phenols on lymphomonocyte cytosolic calcium. J Nutr Biochem. 2005;16:109–113.
- Schaffer S, Podstawa M, Visioli F, et al Hydroxytyrosol-rich olive mill wastewater extract protects brain cells in vitro and ex vivo. J Agric Food Chem. 2007;55:5043–5049.
- Fabiani R, De Bartolomeo A, Rosignoli P, et al Virgin olive oil phenols inhibits proliferation of human promyelocytic leukemia cells (HL60) by inducing apoptosis and differentiation. J Nutr. 2006;136:614–619.
- Guichard C, Pedruzzi E, Fay M, et al Dihydroxyphenylethanol induces apoptosis by activating serine/threonine protein phosphatase PP2A and promotes the endoplasmic reticulum stress response in human colon carcinoma cells. Carcinogenesis. 2006;27:1812–1827.
- Takeda Y, Bui VN, Iwasaki K, et al. Influence of olive-derived hydroxytyrosol on the toll-like receptor 4-dependent inflammatory response of mouse peritoneal macrophages. Biochem Biophys Res Commun. 2014;446:1225–1230.
- Funakoshi-Tago M, Sakata T, Fujiwara S, et al Hydroxytyrosol butyrate inhibits 6-OHDA-induced apoptosis through activation of the Nrf2/HO-1 axis in SH-SY5Y cells. Eur J Pharmacol. 2018;834:246–256.
- Niino T, Tago K, Yasuda D, et al A 5-hydroxyoxindole derivative attenuates LPS induced inflammatory responses by activating the p38-Nrf2 signaling axis. Biochem Pharmacol. 2018;155:182–197.
- Trujillo M, Mateos R, Dollantes de Teran L, et al Lipophilic hydroxytyrosyl esters. Antioxidant activity in lipid matrices and biological systems. J Agric Food Chem. 2006;54:3779–3785.
- Buisman GJH, Ctw VH, Kramer GFH, et al Enzymatic esterifications of functionalized phenols for the synthesis of lipophilic antioxidants. Biotechnol Lett. 1998;20:131–136.
- Torres de Pinedo A, Peñalver P, Pérez-Victoria I, et al. Synthesis of new phenolic fatty acid esters and their evaluation as lipophilic antioxidants in an oil matrix. Food Chem. 2007;105:657–665.
- Bernini R, Mincione E, Barontini M, et al Convenient synthesis of hydroxytyrosol and its lipophilic derivatives from tyrosol or homovanillyl alcohol. J Agric Food Chem. 2008;56:8897–8904.
- Zhou D-Y, Sun Y-X SF. Preparation and antioxidant activity of tyrosol and hydroxytyrosol esters. J Funct Foods. 2017;37:66–73.
- Honda T, Kubo S, Masuda T, et al. Synthesis and in vivo influenza virus-inhibitory effect of ester prodrug of 4-guanidino-7-O-methyl-Neu5Ac2en. Bioorg Med Chem Lett. 2009;19:2938–2940.
- Nakazawa Y, Pauze M, Fukuyama K, et al Effect of hesperetin derivatives on the development of selenite-induced cataracts in rats. Mol Med Rep. 2018;18:1043–1050.
- Torres de Pinedo A, Peñalver P, Rondón D, et al. Efficient lipase-catalyzed synthesis of new lipid antioxidants on a catechol structure. Tetrahedron. 2005;61:7654–7660.
- Grasso S, Siracusa L, Spatafora C, et al Hydroxytyrosol lipophilic analogues: enzymatic synthesis, radical scavenging activity and DNA oxidative damage protection. Bioorg Chem. 2007;35:137–152.
- Capasso R, Cristinzio G, Evidente A, et al Isolation, spectroscopy and selective phytotoxic effects of polyphenols from vegetable waste waters. Phytochemistry. 1992;31:4125–4128.
- DellaGreca M, Previtera L, Temussi F, et al Low-molecular-weight components of olive oil mill waste-waters. Phytochem Anal. 2004;15:184–188.
- Allouche N, Fki I, Sayadi S. Toward a high yield recovery of antioxidants and purified hydroxytyrosol from olive mill wastewaters. J Agric Food Chem. 2004;52:267–273.
- Capasso R, Evidente A, Avolio S, et al A Highly convenient synthesis of hydroxytyrosol and its recovery from agricultural waste waters. J Agric Food Chem. 1999;47:1745–1748.
- Deffieux D, Gossart P, Quideau S. Facile and sustainable synthesis of the natural antioxidant hydroxytyrosol. Tetrahedron Lett. 2014;55:2455–2458.
- Bovicelli P, Antonioletti R, Mancini S, et al Expedient synthesis of hydroxytyrosol and its esters. Synth Commun. 2007;37:4245–4252.
- Bernini R, Mincione E, Crisante F, et al. A novel use of recyclable polymer-supported IBX: an efficient chemoselective and regioselective oxidation of phenolic compounds. The case of hydroxytyrosol derivatives. Tetrahedron Lett. 2009;50:1307–1310.
- Fernández O, Tenllado D, Martín D, et al Immobilized lipases from Candida antarctica for producing tyrosyl oleate in solvent-free medium. Biocat Biotrans. 2012;30:245–254.
- Pezzella A, Lista L, Napolitano A, et al An expedient one-pot entry to catecholestrogens and other catechol compounds via IBX-mediated phenolic oxygenation. Tetrahedron Lett. 2005;46:3541–3544.