
ABSTRACT
Ethyl (R)-2-benzyloxy-2-isopropylhydrogenmalonate is a key intermediate for the synthesis of the side chain in ergopeptines. In this work, we adopted a method to prepare enantiomerically pure title monoester via immobilized Candida antarctica lipase B (Novozym 435)-catalyzed hydrolysis of the corresponding diester.
Graphical Abstract
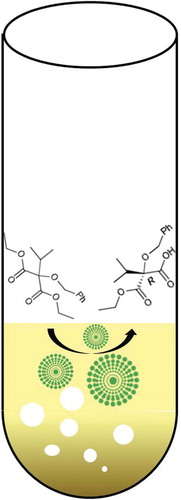
Novozyme 435 catalyzed hydrolysis reaction to prepare the key intermediate for the synthesis of peptide side chain of ergopeptides.
Ergot alkaloids (EA) are the metabolic products of the parasitic fungus Claviceps. In early history of EA, they have caused mass poisoning of animals and humans for their extensive and powerful pharmacological activities [Citation1,Citation2]. The structural characteristic of natural EA is the tetracyclic ergoline ring methylated on nitrogen N-6 and variously substituted on C-8. Depending on the type of R substituent on C-8 of the ergoline ring, they can be divided into four groups: clavine alkaloids and 6,7-secoergolenes, simple lysergic acid derivatives, peptide alkaloids-cyclol ergot alkaloids (CEA), lactam ergot alkaloids (LEA) [Citation3]. Among them, peptide ergot alkaloids (ergopeptines) are a predominant class of EA that have a variety of pharmaceutical activities, such as anti-cerebrovascular insufficiency [Citation3], prolactin inhibition [Citation4], anti-parkinsonism [Citation5], treatment of migraine [Citation6],etc.
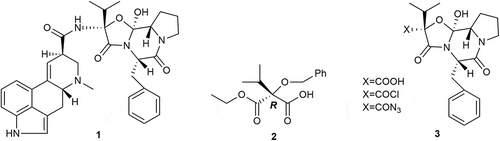
Actually, ergopeptines are believed to be the most important natural pharmaceuticals and toxins in human history [Citation7], which are composed of lysergic acid and some tripeptides [Citation8]. In the synthesis of ergopeptides (eg. ergocristine 1), the interesting peptide side chain has been a challenge that remained to be addressed, while 2 is the key intermediate for the synthesis of peptide side chain intermediates 3 (). At present, the optical isomer of compound 2 is commonly obtained through tedious chemical resolution, using (R)-(+)-pseudoephedrine hydrochloride [Citation9] or D-(-)-threo-1-(p-nitrophenyl)-2-amino-1,3-propanediol [Citation10] as a chiral resolving agent. Alternatively, biocatalysts have proved to be a powerful tool for the preparation of enantiomerically enriched compounds, even if the sterically hindered substrates [Citation11,Citation12]. We herein first report a highly enantioselective approach to obtain 2 by commercially available enzymes under mild and convenient conditions. We planned a chemoenzymatic route from 4 to 2 ().
Scheme 1. Structures of ergocristine 1, (R)-monoester 2 and important intermediates 3.
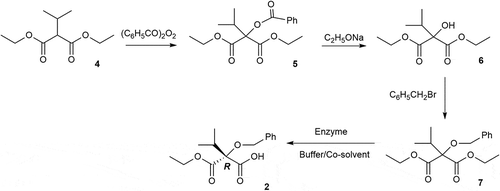
Scheme 2. Preparation of 2 via a chemoenzymatic route.
Materials and methods
Chemicals
Novozym 435 (lipase B from Candida antarctica, immobilized on a microporous acrylic resin, 10,000 PLU/g), Lipase AS “Amano” (43,106) from Aspergillus niger (12,000 U/g), Lipase G (43,109) from Penicillium camemberti (50,000 U/g), Lipase M (43,110) from Mucor javanicus (50,000 U/g), Lipozyme RM IM (43111M) from Rhizomucor miehei (250 IUN/g), Lipozyme TL IM (43112M) from Thermomyces lanuginosus (250 IUN/g), and Lipase PS (Burkholderia cepacia lipase, 20 U/mg).
Synthesis of compound diethyl 2-(benzoyloxy)-2-isopropylmalonate (5)
To a suspension of NaH (4.80 g, 0.120 mmol, 60% oil dispersion) in anhydrous toluene (150 mL). The mixture was stirred well and cooled to 15 °C, 4 (20.2 g, 0.100 mmol) was added dropwise. Then the reaction mixture was stirred at 50 °C for 1 h. After no obvious bubbles released, the reaction mixture was cooled to 5 °C. A solution of benzoyl peroxide (24.2 g, 0.100 mmol) dissolved in anhydrous toluene (200 mL) was added dropwise via an addition funnel maintaining temperature below 10 °C. After the completion of the addition, the reaction mixture was stirred at 40 °C for 1 h. Activated carbon (2.00 g) was added in batches under ice-bath and then the mixture was stirred at 40 °C for 1 h and stirred at 20 °C overnight. The reaction mixture was filtered and the wet cake was washed with toluene and water, the filtrate was collected together and washed to neutral with water, the organic phase was dried over anhydrous sodium sulfate and concentrated. Then the residue was distilled under reduced pressure at 80 °C, 26 Pa to obtain 29.0 g crude product. The material was ready for the next step without purification. 1H NMR (400 MHz, CDCl3): δ 8.10–8.06 (2H, m), 7.57 (1H, t, J = 7.5 Hz), 7.46 (2H, t, J = 7.7 Hz), 4.28 (2H, q, J = 7.1 Hz), 2.69–2.56 (1H, m), 1.27 (6H, t, J = 7.1 Hz), 1.2 (6H, d, J = 6.9 Hz).
Synthesis of compound diethyl 2-hydroxy-2-isopropylmalonate (6)
5 (14.4 g, 0.045 mmol) was added to 20% sodium ethoxide solution (16.7 mL) at 35–40 °C and keep stirring for 8 min at this temperature. The reaction mixture was cooled to 15 °C, acetic acid (2.88 g, 0.048 mmol) was added to quench the excess sodium ethoxide, water (100 mL) was added to the mixture and then the mixture was extracted with petroleum ether (3 × 50 mL), the organic phase was washed successively with saturated sodium carbonate and water, dried over anhydrous sodium sulfate, the solvent was evaporated under reduced pressure. The residue was distilled under reduced pressure at 19 Pa, and the fraction of 70–100 °C was collected as ethyl benzoate and 6. The mixture was separated by a silica column to give apple-like odor colorless oil 6 6.4 g, 65.3% yield. 1H NMR (400 MHz, CDCl3): δ 4.27 (4H, q, J = 5.8 Hz), 2.72–2.58 (1H, m), 3.69 (1H, s), 1.30 (6H, t, J = 7.1 Hz), 0.93 (6H, d, J = 6.8 Hz).
Synthesis of compound diethyl 2-(benzyloxy)-2-isopropylmalonate (7)
To a suspension of NaH (0.8 g, 0.033 mol, 60% oil dispersion) in anhydrous N,N-dimethylformamide (12.5 mL), then diester 6 (3.4 g, 0.016 mmol) was added dropwise under ice-bath. When no gas was generated during the reaction, the reaction mixture was heated to 70 °C, and benzyl bromide (3.20 g, 0.019 mol) was added dropwise. After that, the mixture was stirred at 75 °C for 2 h. Then ethanol (2.00 mL) was added and stirred at 75 °C for another 30 min to quench the excess benzyl bromide. The reaction mixture was neutralized with acetic acid after cooling to room temperature, diluted with water (150 mL), then extracted with petroleum ether (3 × 70 mL), the organic phase was washed successively with saturated sodium carbonate and water, dried over anhydrous sodium sulfate, filtrated, and solvent was removed under reduced pressure. The crude residue was purified on a silica gel column to afford the pure 7 as a yellow oil 3.6 g, 80.3% yield and the purity was 98% by gas chromatography. 1H NMR (400 MHz, CDCl3): δ 7.40 (2H, d, J = 7.3 Hz), 7.36–7.23 (3H, m), 4.75 (2H, s), 4.26 (4H, q, J = 7.1 Hz), 2.56–2.49 (1H, m), 1.29 (6H, t, J = 7.1 Hz), 1.05, (6H, d, J = 6.9 Hz).
General procedure for lipase-catalyzed hydrolysis of diethyl 2-(benzyloxy)-2-isopropylmalonate (7)
7 (0.2 mmol) was dissolved in 0.3 ml co-solvent and then this solution was added to 0.7 ml phosphate buffer with 20 mg lipase. The mixtures were continuously stirred at 20–60 °C for 24 h. After the reactions were stopped, adjusting the pH to 2.0 with 1 M HCl and then the mixtures was extracted with ethyl acetate (3 × 2 ml). The extracts were dried over anhydrous magnesium sulfate and analyzed by chiral HPLC. 1H NMR (400 MHz, CDCl3): δ 7.42–7.26 (5H, m), 4.76 (1H, d, J = 10.9 Hz), 4.57 (1H, d, J = 10.9 Hz), 4.32 (2H, q, J = 7.2 Hz), 2.56–2.42 (1H, m), 1.32 (3H, t, J = 7.2 Hz), 1.08, (6H, dd, J = 3.3, 6.9 Hz); HRMS (ESI) m/z: [M+ Na]+ calcd for C15H20O5Na 303.1208, found 303.1203.
High performance liquid chromatography (HPLC) analysis
The determination of enantiomeric excess (ee) of 2 was performed by chiral HPLC analysis carried out on a HPLC system equipped with UV wavelength detector and autosampler, using a Chiralcel IA-3 column (25 × 4.6 mm) thermostated at a temperature of 40 °C. The mobile phase was a mixture of hexane, isopropyl alcohol, ethanol and trifluoroacetic acid (70:15:15:0.1, v/v/v/v) at a flow rate of 1.0 ml/min and UV detection was performed at 293 nm. The yield of 2 was measured by using the following equation: yield (%) = Mp/MS, Mp and MS are the final mole of 2 and the original mole of the substrate, respectively. The ee were obtained by the ratio of peak areas of 2 (A1) and (S)-enantiomer (A2) as follows: ee (%) = (A1 − A2)/(A1 + A2) × 100.
Results and discussion
7 as enzyme catalyzed substrate was successfully synthesized from commercially available 4 with three-step chemical transformations (). We subsequently focused on the synthesis of 2 by an enzyme-catalyzed desymmetrization procedure starting from the prochiral ester 7. Various lipases were employed for this transformation. More importantly, the use of organic co-solvent has proven to be an effective strategy to overcome the low solubility of hydrophobic substrates in an aqueous medium. Besides, it was reported that enantioselectivity and catalytic activity of enzymes can be improved by organic co-solvent which can optimize the properties of a reaction medium [Citation13–Citation18]. The temperature and pH are also important parameters affecting enzyme activity [Citation19]. Therefore, all above mentioned factors that may affect the enzymatic desymmetrization procedure were investigated in order to obtain the optimum results.
Synthesis of diethyl 2-(benzyloxy)-2-isopropylmalonate (7)
For the preparation of 7, commercially available 4 was chosen as starting material, which was oxidated by dibenzoyl peroxide to give 5. Then the 6 was obtained by alcoholysis of 5. 7 was synthesized by the reaction of 6 and benzyl bromide. We found that the temperature and the content of H2O were the crucial factor of the synthesis of 5 and 6.
Enzyme screening
After the substrate in hand, we started the enzymatic hydrolysis study from enzyme screening. The lipases were used to catalyze the hydrolysis of 7 in phosphate buffer (0.1 M, pH 8.0), and acetone was used as co-solvent in experiments. To our delight, these lipases gave satisfactory results except TL IM and lipases PS (). Lipase AS, Lipase G, Lipase M and RM IM led to moderate activity. However, Novozym 435 showed the excellent enantioselectivity and good yield, with enantiomer being preferentially formed at room temperature. Novozym 435 indeed has a wide range of applications in biocatalytic organic synthesis [Citation20–Citation22]. Thus, Novozym 435 was consequently used throughout the further studies.
Table 1. Various lipases for hydrolysis of 7.
Choice of organic co-solvent
In order to resolve the poor solubility of prochiral substrate 7 in aqueous medium, organic solvent was used as co-solvent to improve the dissolubility and dissolution rate. Besides, the activity and enantioselectivity of Novozym 435 are also influenced by different organic co-solvent. Therefore, we examined various organic co-solvents on the reaction process. As shown in , dimethyl sulfoxide (DMSO) gave the best results, so that DMSO was chosen as co-solvent in the consequent experiments.
Table 2. Effect of co-solvent on hydrolysis of 7.
Effect of concentration of co-solvent
Generally, the concentration of co-solvent also has great influence on the properties of a reaction medium that can affect the enantioselectivity in the process of hydrolysis. Taking into consideration of this, we explored the effect of co-solvent concentration on yield and ee of 2 (Fig. S1). When the concentration of DMSO was raised from 10 to 30%, the desired products were obtained with obviously increased yield and ee. It can be explained by the more solubility of substrate, the better selectivity and reactivity of enzyme. However, when the concentration of DMSO exceeds 30% (v/v), both of selectivity and reactivity were decreased as DMSO increased. This may be due to dilute the substrate concentration in the organic phase, the less contact between substrate and enzyme in the two-phase system. The highest ee was obtained when DMSO concentration was 30% (v/v), indicating that 30% (v/v) of DMSO produced the best effect on the activity and enantioselectivity.
Effect of reaction temperature
It was generally acknowledged that one of the most important limiting factors for industrial applications is the thermal stability of biocatalysts. Fortunately, Novozym 435 was considered to have considerable thermal stability. Therefore, the temperature we examined in present work was broadened from 20 to 60 °C. As shown in Fig. S2, the maximum value of the activity and enantioselectivity were observed at 30 °C and the enzyme was still very active at this temperature. The excessive high temperature led to the conformational change of the active site, which determines the enantioselectivity of Novozym 435. As shown in Fig. S2, the enantioselectivity decreased sharply at temperatures exceeding 30 °C, and 30 °C was selected as the optimum temperature by considering a compromise between yield and ee of 2.
Effect of buffer pH
Similarly, both the activity and enantioselectivity of enzymes are highly influenced by buffer pH [Citation23,Citation24]. The different buffered solutions in the range of pH 5.0 to 9.0 were selected to investigate the effect of buffer pH on the catalyzed reaction (Fig. S3). The results were indicative that Novozym 435 showed higher activity in the buffered solutions of pH 7.0, and declined to different extents beyond this range, indicating that the enzymatic activity was partially inhibited in acidic and strong basic solutions. According to the above experiments, pH 7.0 was the optimum for this hydrolytic reaction.
Effect of substrate concentration and enzyme loading
In organic solvents, enzymes must have a certain amount of water to maintain their catalytic activity [Citation25,Citation26]. During the hydrolysis process, we observed that the yield and ee value raised with the increase of enzyme loading, when the enzyme loading was less than 20 mg. However when the enzyme loading exceeded 20 mg, both yield and ee value showed a downward trend (Fig. S4). It may be that as the enzyme loading increased, the relative water content required to maintain enzyme activity was reduced, resulting in a reduced ability of the enzyme to catalyze hydrolysis.
Effect of reaction time
As shown in Fig. S5, The ee value hardly changed, indicating that lipase had maintained good enantioselectivity during hydrolysis process. However, the yield changed regularly with time. The yield increased rapidly within 3–12 h, and the tendency of transformation became smoothly after the reaction exceeded 24 h. Therefore, 24 h was the optimum for the enzymatic hydrolysis of 7.
Conclusion
In conclusion, chemo-enzymatic strategy is clearly a useful synthetic tool which found its place in the synthesis useful intermediates. We have demonstrated the versatility and efficiency of readily available lipase Novozym 435 for enantioselective synthesis of 2, which is the key intermediate for the synthesis of peptide side chain of ergopeptides. The present enzyme-catalyzed procedure is very mild resulting in the desired product in excellent ee and yield. Specifically, 99% ee and 50% yield could be reached via Novozym 435 (20 mg) hydrolysis of 0.2 mmol substrate 7 in 1.0 ml phosphate buffer (pH 7.0)/DMSO (30% (v/v)) solvent system at 30 °C for 24 h.
Authors contribution
Qun Lu developed the ideas. Weizhen Fang and Yu Zhao designed the experiments. Jing Ding carried out all experiments and wrote the paper. Zhijun Yang discussed and analyzed the data.
Supporting_information.docx
Download MS Word (12.2 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Sinz A. Die Bedeutung der Mutterkorn‐Alkaloide als Arzneistoffe. Weit mehr als nur α‐Blocker. Pharm Unserer Zeit. 2008;37:306–309.
- Somei M, Yokoyama Y, Murakami Y, et al. Recent synthetic studies on the Ergot alkaloids and related compounds. J Cheminform. 2001;32:191–257.
- Flieger M, Wurst M, Shelby R. Ergot alkaloids-sources, structures and analytical methods. Folia Microbiol. 1997;42:3–30.
- Lee MR. The history of ergot of rye (Claviceps purpurea) III: 1940-80. JR Coll Physicians Edinburgh. 2010;40:77–80.
- Parkes JD, Marsden CD, Donaldson I, et al. Bromocriptine treatment in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1976;39:184–193.
- Eadie MJ. Ergot of rye-the first specific for migraine. J Clin Neurosci. 2004;11:4–7.
- Schiff PL Jr. Ergot and its alkaloids. Am J Pharm Educ. 2006;70:98.
- Cheng JZ. Nature’s approach toward ring formation and structural diversity in ergot alkaloid biosynthesis. Doctoral dissertation, Massachusetts Institute of Technology, (2011).
- Stadler PA, Guttmann S, Hauth H, et al. Die Synthese der Alkaloide der Ergotoxin-Gruppe. 70. Mitteilung über Mutterkornalkaloide [1]. Helv Chim Acta. 1969;52:1549–1564.
- Moldvai I, Temesvári-Major E, Incze M, et al. Enantioefficient synthesis of α-ergocryptine: first direct synthesis of (+)-lysergic acid. J Org Chem. 2004;69:5993–6000.
- Pogorevc M, Faber K. Biocatalytic resolution of stereically hindered alcohols, carboxylic acids and esters containing fully substituted chiral centers by hydrolytic enzymes. J Mol Catal B. 2000;10:357–376.
- Sugai T, Higashibayashi S, Hanaya K. Recent examples of the use of biocatalysts with high accessibility and availability in natural product synthesis. Tetrahedron. 2018;74:3469–3487.
- Hansen TV, Waagen V, Partali V, et al. Co-solvent enhancement of enantioselectivity in lipase-catalysed hydrolysis of racemic esters. A process for production of homochiral C-3 building blocks using lipase B from Candida antarctica. Tetrahedron: Asymmetry. 1995;6:499–504.
- Kato K, Gong Y, Saito T, et al. Efficient preparation of optically active ketoprofen by Mucor javanicus lipase immobilized on an inorganic support. J Biosci Bioeng. 2000;90:332–334.
- Zhang LQ, Zhang YD, Xu L, et al. Lipase-catalyzed synthesis of RGD diamide in aqueous water-miscible organic solvents. Enzyme Microb Tech. 2001;29:129–135.
- Chen Y, Xu JH, Pan J, et al. Catalytic resolution of (RS)-HMPC acetate by immobilized cells of Acinetobacter sp. CGMCC 0789 in a medium with organic cosolvent. J Mol Catal B. 2004;30:203–208.
- Fernández-Lorente G, Palomo JM, Mateo C, et al. Fernandez-Lafuente, R. Resolution of paroxetine precursor using different lipases: influence of the reaction conditions on the enantioselectivity of lipases. Enzyme Microb Tech. 2004;34:264–269.
- Zhang W, Ni Y, Sun Z, et al. Biocatalytic synthesis of ethyl (R)-2-hydroxy-4-phenylbutyrate with Candida krusei SW2026: a practical process for high enantiopurity and product titer. Process Biochem. 2009;44:1270–1275.
- Bansode SR, Hardikar MA, Rathod VK. Evaluation of reaction parameters and kinetic modelling for Novozym 435 catalysed synthesis of isoamyl butyrate. J Chem Technol Biotechnol. 2017;92:1306–1314.
- Villalba M, Verdasco-Martín CM, Dos Santos JC, et al. Operational stabilities of different chemical derivatives of Novozym 435 in an alcoholysis reaction. Enzyme Microb Tech. 2016;90:35–44.
- Polloni AE, Chiaradia V, Figura EM, et al. Polyesters from macrolactones using commercial lipase NS 88011 and Novozym 435 as Biocatalysts. Appl Biochem Biotech. 2018;184:659–672.
- Karlsson S. Development of an Enantioselective Novozym 435 mediated acetylation for the preparation of (1 S, 3 R)-3-Acetamidocyclohexane-1-carboxylic acid. Org Process Res Dev. 2016;20:1336–1340.
- Dong HP, Wang YJ, Zheng YG. Enantioselective hydrolysis of diethyl 3-hydroxyglutarate to ethyl (S)-3-hydroxyglutarate by immobilized Candida antarctica lipase B. J Mol Catal B. 2010;66:90–94.
- Long WS, Kow PC, Kamaruddin AH, et al. Comparison of kinetic resolution between two racemic ibuprofen esters in an enzymic membrane reactor. Process Biochem. 2005;40:2417–2425.
- Basheer S, Mogi K, Nakajima M. Surfactant‐modified lipase for the catalysis of the interesterification of triglycerides and fatty acids. Biotechnol Bioeng. 1995;45:187–195.
- Zaks A, Klibanov A,M. Enzymatic catalysis in nonaqueous solvents. J Biol Chem. 1988;263:3194–3201.