
ABSTRACT
In this report, an artificial antigen (PFLX–BSA: Pefloxacin connected bovine serum albumin) was successfully prepared. The monoclonal antibody against pefloxacin was produced and characterized using a direct competitive ELISA. The linear range of detection was 0.115–6.564 µg/L. The limit of detection defined as IC15 was 0.170 ± 0.05 µg/L and the IC50 was 0.902 ± 0.03 µg/L. The antibody variable region genes were amplified, assembled, and sequenced. A three–dimensional structural model of the variable region was constructed to study the mechanism of antibody recognition using molecular docking analysis. Three predicted essential amino acids, Thr53, Arg97 of heavy chain and Thr52 of light chain, were mutated to verify the theoretical model. Three mutants lost binding activity significantly against pefloxacin as predicted. These may provide useful insights for studying antigen–antibody interaction mechanisms to improve antibody affinity maturation in vitro.
Graphical Abstract
The dc-ELISA of IC15 was 0.151±0.07 ng/mL and IC50 was 0.891±0.03 ng/mL. Molecular docking analysis indicated that the Ser52, Thr53, Arg97, Tyr100, Leu51, Thr52, Ser53 amino acid residues played the decisive roles during recognition.
Pefloxacin (PFLX) is an antibacterial drug that belongs to the fluoroquinolone class. It has been widely used in the veterinary industry to treat and prevent various infectious diseases. The residues of these drugs entering the food chain present a potential danger such as toxic effect (including central nervous system toxicity, phototoxicity, cardiotoxicity, arthropathy, and tendon toxicity), resistant strain of bacteria, allergic reaction, etc, which are harmful to human health [Citation1,Citation2]. As a result, PFLX should be monitored and controlled strictly.
Governmental agencies have set limitations on the levels of residues in recent years. Several traditional methods for detection of PFLX residues,such as spectrophotometry [Citation3], HPLC [Citation4], capillary electrophoresis and microbiological assay [Citation5,Citation6]. These methods are well-proven and widely accepted, but they are often viewed as laborious and time intensive in samples pretreatment, moreover, a significant investment in equipment. Immunoassays, which are based on antigen-antibody interaction, can avoid the drawbacks of chromatographic techniques. They are extremely effective for monitoring and detecting hazardous residues in food and environment samples [Citation7,Citation8]. There have been a few researchers developing ELISA method to detect PFLX. However, most of these studies used either polyclonal antibody (pAb) [Citation9,Citation10] or a broad specific monoclonal antibody (mAb) [Citation11,Citation12], which simultaneously detects multifarious fluoroquinolones; however, the sensitivity against PFLX is limited. In this study, a monoclonal antibody against PFLX with high specificity was produced and a direct competitive ELISA was established to detect PFLX based on this monoclonal antibody.
To obtain a high serum titer antibody
The key step is the design of a hapten. The mechanism of PFLX recognition by the antibody is yet to be fully elucidated because crystallographic studies can provide only limited information on their ternary complex structures. Hapten designs in the methods quoted and in immunoassays are still being developed based on trial and error. This has been restriced immunoassay development. In recent years, with the progress of computational biology, it has been possible to study the protein ligand interaction on silicon. As we know, antibodies possess several highly conserved framework regions (FR) and complementarity-determining regions (CDR), but the folds adopted by most of CDR loops are restricted to few main chain conformations, which called canonical structures [Citation13]. A large number of crystal structures of antibody are now available in PDB bank, so these crystal structures could be used as templates to modeling a target antibody. Previous researches had confirmed that the homology modeling of the protein could be built with a reasonably high level of confidence [Citation14]. The binding conformation and binding interactions of the ligand with the antibody could be modeled by docking programs when structures are available. Several successful antibody affinity maturation have been reported based on artificial intelligent design, a promising, fast and efficient technique to enhance the affinities of antibodies in vitro [Citation15].
Materials and methods
Materials
Pefloxacin was provided by China Institute of Veterinary Drugs Control (Beijing, China). PFLX-BSA (BSA: Albumin from bovine serum) as immunogen and PFLX-OVA (albumin) as coating antigen were conjugated in our laboratory. FCA (Freund’s complete adjuvant), FIA (Freund’s incomplete adjuvant) and GaMIgG-HRP were obtained from Simga-Aldrich Company(Shanghai, China). PEG (Polyethylene glycol) and o-phenylenediamine (OPD) were also from Simga-Aldrich Company. primer of heavy chain (VH) and light chain (VL) [Citation16] were synthesized by Sangon (Shanghai, China). RNeasy Mini Kit and DNA gel recovery kit were purchased from Omega Bio-Tek Company (Guangzhou, China). GoScript reverese transcription system was purchased from Promega Company (Beijing, China). The pLIP6/GN vector used for soluble protein expression was generously provided by Dr. Frédéric Ducancel (Pharmacology and Immunoanalysis Department, CEA/Saclay, Gif-sur-Yvette, France), Which include Alkaline phosphatase (AP) genes.
Preparation of the PFLX complete antigen
To prepare complete antigen, PFLX was connected to the vector protein by the chemical reaction method. First, 13.3 mg of PFLX was added to 3 mL of anhydrous DMF (N,N–dimethylformamide) containing 21.2 mg of NHS (N–hydroxysuccinimide) and 70.5 mg of EDC [1–(3–dimethylaminopropyl)–3–ethylcarbodiimide]. The mixture was stirred at room temperature overnight and then centrifuged to remove precipitated urea. The resulting active ester was added slowly to 5 mL of protein solution (50 mg BSA as the immunogen in 0.01 M phosphate buffered saline, PBS, pH 7.4). The reaction mixture was stirred at 4°C for 3 h and then dialyzed against 0.01 M PBS for 3 days.
Immunization, fusion, and production of a monoclonal antibody
Immunized 8–week–old BALB/c mice (Military Academy of Medical Science Beijing, China) were injected intraperitoneally into PFLX–BSA (100 μg in 0.25 mL PBS), mixed with an equal volume of FCA to form an emulsion. Subsequently, FIA substituted the FCA as the emulsifier and mice were injected with 100 μg of immunogen in booster injections every 3 weeks. After the third booster immunization, blood was collected from the mice tail vein, and the serum titer was determined to compete with PFLX, and continued until the serum titer was >1:8,000. The mice were sacrificed 3 days later and their spleens were harvested. Fusion experiments were carried out as described previously. The positive hybridoma cells were subcloned by a limiting dilution method in the presence of thymocytes of BALB/c mice as feeder cells, according to standard protocols [Citation17,Citation18].
Ascites fluid was produced in pristine-primed BALB/c mice by intraperitoneal injection of 1.6 × 10 [Citation6] hybridoma cells. The fluid was collected 7–10 days after hybridoma injection. Ascites fluid was initially purified by caprylic/ammoniunl sulfate precipitation, and then purified using a protein A–agarose affinity chromatography column according to the manufacturer’s procedure.
Development of dc-ELISA for PFLX
Ninety–six–well polystyrene ELISA plates were coated with 0.05 μg per well of the monoclonal antibody (mAb) solution by overnight incubation at 4°C. Coated plates were washed three times with a PBST solution and the wells were blocked with 100 μL per well of 1% BSA for 1 h at 37°C, 50 uL of analyte standards in 10% (v/v) methanol in PBS and 50 μL of enzyme tracer PFLX–HRP solution (1:8,000 in PBS) were added to the wells. After 1 h incubation at 37°C the plates were washed and 100 μL per well of substrate solution was added. The absorbance of the solutions in the wells was measured at 450 nm after incubation of the reaction for 20 min at room temperature.
Spiking of milk samples
Milk samples (Supermarket, Tianjin, China) were chosen for the spiking study. Selected one milk was used as the blank sample in this study and the other milk samples were spiked with an PFLX standard solution (0.5, 1.0, 2.5, 5.0, 7.5, and 10.0 µg/L). For ELISA, 1 mL PFLX of milk sample and 2 mL of 7.5% (w/v) trichloroacetic acid aquatic solution were added to a 5 mL centrifuge tube. The sample was thoroughly mixed and centrifuged at 5,000 g for 30 min to deproteinate. The upper fat layer was discarded, the supernatant was diluted using 0.01 M PBS (pH 7.4) and then used for detection.
Cloning of VL and VH genes and the scFv gene assembly
The total RNA was extracted from the mAb hybridoma cell line (~1 × 107 cells) using the RNeasy mini kit according to the manufacturer’s procedure. The first–strand cDNA was synthesized from RNA using a reverse–transcription PCR kit, then cDNA as the template was used to amplify the heavy and light chains of the antibody variable region using primers VL–back, VL–forward, VH–back, and VH–forward. After purification and quantification, the collected VH and VL DNAs were subjected to PCR splicing by overlap extension to assemble the scFv gene with a (Gly4Ser)3 linker.
Expression and identification of the anti–PFLX scFV–AP
The anti–PFLX–scFv PCR product and pLIP6/GN vector [Citation19] were digested using SfiI and NotI restriction enzymes. After the ligation reaction, the recombinants were transformed into E. coli JM109 competent cells, and positive colonies were used for both restriction enzyme analysis and sequence analysis. Sequences were determined commercially (Genewiz Biological Technology Co. Ltd., Beijing, China).
The recombinant plasmid (scFv–PLIP6/GN) was extracted and transformed into E. coli BL21 competent cells. The colony was cultured in LB (Luria–Bertani) medium containing 100 mg/L ampicillin and IPTG (2 mmol/L) was added to the medium when the OD600 reached 0.8–1.0 to induce the production of the soluble scFv. The culture was further incubated at 28°C with shaking at 180 rpm overnight, followed by centrifugation at 5,000 × g for 19 min, and the anti–PFLX scFv–AP present in the periplasm was extracted using the cold osmotic shock method[Citation18]. The supernatant was analyzed for the presence of soluble scFv–AP by SDS–PAGE.
Activity analysis of anti-PFLX scFv-AP
Microplates were coated with 0.4 µg per well of PFLX–OVA overnight at 37°C. After the plates were washed twice with PBS containing 0.1% Tween–20 (PBST), the wells were then blocked with 0.5% skim milk. After the blocking buffer was discarded, the scFv–AP were added to the wells and incubated at 37°C for 1 h. The plates were then washed five times with PBST and 100 µL per well of AP–substrate (1 mmol/L p–nitrophenyl phosphate, 1 mol/L Tris–HCl, 10 mmol/L MgCl2, 50 mmol/L ZnCl2; pH 8.0) was added. After incubation for 20 min at 37°C, the reaction was stopped with 3 mol/L NaOH and the absorbance was measured at 405 nm.
Homology model building and validation
Swiss–Model Workspace was used to identify a suitable template for modeling. A 3D structural model of the variable regions of the antibody was then generated using Modeller9v2 on the basis of the structure of the template. The model was optimized by adding hydrogen atoms at pH 7.0 and then using the CHARMM all–atom force field minimization with the implicit solvent model set to none. The energy was minimized for at least 1000 steps until the gradient converged to 0.5 kcal mol−[Citation1] using the steepest descent protocol available in Discovery Studio (DS) to remove any steric clashes and to stabilize the models. The secondary structure of the model was analyzed by the SOMPA Program (http://swissmodel.expasy.org/).
A highly reliable model was quite essential. PROCHECK is an efficient tools for evaluating variable region 3D model quality. PROCHECK checked for valid stereochemistry which produces a number of PostScript plots analysing its overall and residue-by-residue geometry.
Antigen–antibody docking
To map the probable epitope, the DS program was used to dock PFLX into the active site of the variable region of the antibody. DS is a fast automated docking program that is currently considered one of the most reliable bioinformatics tools. Libdock was used as the docking mode and the PFLX molecule was drawn by ChemDraw3D. All water molecules from the model structure were removed and hydrogen atoms were added. The active site for docking was defined as all atoms within 6.5 Å radius of the co–crystallized ligand. The default parameters in the DS program module were used.
Point mutation of scFv and validation docking
To evaluate the accuracy of the computer model of docking, three amino acids, Thr53, Arg97 and Thr52, which contribute most to the binding were mutated to Alanine. Oligonucleotide-directed mutagenesis was performed by over-lap PCR as the screened scFv template with the mutant primers (Primers scFvThr53-F: 5′-TCAATTAGCGCAGGTGGTGGCGGCTACTATGCAG-3′ and scFvThr53-R: 5′-CTGCATAGTAGCCGCCACCACCTGCGCTAATTGA-3′ for Thr52 mutant; Primers scFvArg97-F: 5′-TATTGTGCAGCAAGCAATTACTACGGTGGAAGTA-3′ and scFvArg97-R: 5′- TACTTCCACCGTAGTAATTGCTTGCTGCACAATA-3′ for Arg97 mutant; Primers scFvThr52-F: 5′-ATTTATCTCGCATCCAACCTGGCTTCTGGAGTCC-3′ and scFvThr52-R: 5′-GGACTCCAGAAGCCAGGTTGGATGCGAGATAAAT-3′ for Thr52 mutant), and the amplified scFv mutants were inserted into PLIP6/GN vector, respectively. For scFv expression, the constructed mutant vectors were transformed into E. coli BL21 for expression of the mutated scFv antibody, After the target scFv was purified using affinity chromatography according to the manufacturers’ directions. the binding activity of the mutant scFv products was determined by ELISA as wild-type mutated scFv.
Results
Development of dc–ELISA for PFLX
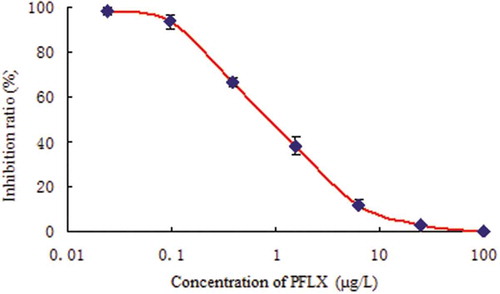
Considering the possible use of the developed ELISA as a future assay, we carried out a dc–ELISA (direct competitive ELISA) experiment. Some parameters of dc–ELISA were optimized, including the quantity of the antibody coating, the concentration of the PFLX product, the competition time, and the concentration of the PFLX–HRP. A standard curve derived from the above experiment is presented in . The linear working range was 0.115–6.564 µg/L. The sensitivity of the dc–ELISA (IC50) was 0.902 µg/L. The limit of detection of dc–ELISA was 0.170 µg/L. The IC50 is lower than the reported ELISA using a fluoroquinolone polyclonal antibody [Citation20,Citation21] and previous monoclonal antibodies [Citation22,Citation23].
Figure 1. Standard curve of PFLX by dc–ELISA.
To study the ability of the mAb to bind to other bactericide analogs, the cross reactions (CRs) of the mAb with PFLX and other bactericide analogs, including marbpefloxacin, fleroxacin, norfloxacin, ofloxacin, enrPefloxacin, enoxacin, ciprPefloxacin, sarafloxacin, difloxacin, and sparfloxacin, were tested by the optimized assay (). The CR values of mAb with other bactericide analogs were below 1%, except for marbPefloxacin (12.3%), ofloxacin (4.7%), and enrPefloxacin (2.5%). Therefore, the mAb could be used for detection PFLX residues in food samples.
Table 1. Cross reactions (CRs) of mAb with PFLX and other bactericide analogs.
Spiking of milk samples
PFLX was added to milk samples at five concentrations (0.5, 1.0, 2.5, 5.0, 7.5, and 10.0 µg/L) and the detection results from the developed assay were validated by HPLC (Table S1). The average recoveries of Pefloxacin by ELISA were in the range of 95–103%. The results showed a good correlation (R2= 0.9960) between the two methods, which indicated that the developed immunoassay was reliable for the detection of PFLX in milk samples.
Cloning of the VH and VL chain genes and the scFv gene assembly
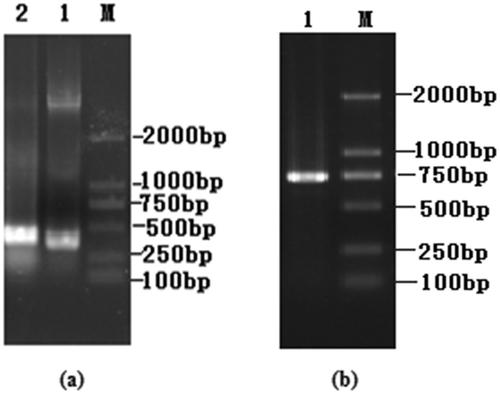
Total RNA from the hybridoma cell G10F5 was used to clone the VH and VL domain cDNA. These two genes encoding the variable domains were amplified by PCR and sequenced. The VH and VL gene products formed bands at ~340 and ~320 bp on an agarose gel ()). The VH and VL fragments containing linker overhangs were assembled by splicing overlap extension PCR to generate the scFv gene (750 bp; )).
Figure 2. Cloning of the VH and VL chain genes and the scFv gene assembly. (a) Agarose gel electrophoresis of VH and VL genes of PFLX. Lane M, DL 2000 DNA marker; Lane 1, VL gene. Lane 2, VH gene. (b) Agarose gel electrophoresis of scFv of PFLX. Lane M, DL 2000 DNA marker; Lane 1, scFv gene.
Expression and identification of the anti–PFLX scFv–AP
The scFv products and vector PLIP6/GN were both digested with SfiI and NotI restriction enzymes. The digested products were purified with a PCR product purification kit and then ligated using the T4 DNA ligation enzyme. The ligation products were transformed into E. coli strain JM109 competent cells and the extracted plasmid was characterized by restriction enzyme digestion and sequence analysis (Fig. S1 Aand Fig. S2).
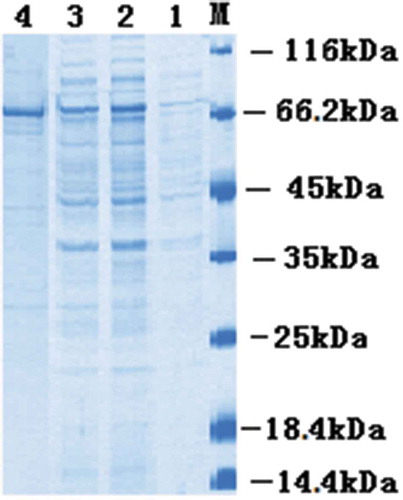
The positive recombinant plasmid was used to transform E. coli BL21 competent cells. When the OD600 reached 0.8–1.0, expression of the target protein was induced by the addition of IPTG and the culture grown overnight at 25°C. The periplasmic protein was extracted by means of cold osmotic shock. As shown in , soluble protein of ~75 kDa was expressed as expected by SDS–PAGE analysis.
Figure 3. SDS–PAGE analysis of the anti–PFLX scFv–AP fusion protein.
Lane M, marker 116 kDa; Lane 1, before induction; Lane 2, after induction; Lane 3, the supernatant; Lane 4, purified scFv-AP.
Activity analysis of anti–PFLX scFv–AP
To identify the activity of anti–PFLX scFv–AP, an indirect ELISA was carried out and each experiment was tested in triplicate using purified anti–PFLX scFv–AP. The result is presented in Fig. S3. With the increasing concentration of scFv–AP, the OD450 gradually became stronger. What’s more, both were dose dependent. This suggest the PFLX antigen can strongly recognized by the scFv.
Molecular modeling of the variable region and validation
In this study, we were interested in determining the variable region 3D structure of an antibody using homology modeling, which was extremely dependent on the identity with template proteins. Templates were identified with the help of the SWISS–MODEL Workspace and a crystal structure (PDB ID: 4GQP) was selected as the final template because of its high-resolution structure (2.0 Å) and 89% sequence identity with the target antibody sequence. The variable region 3D structure of the mAb was generated using Modeller9v2 and was optimized subsequently. The model (blue) is superimposed to that of 4GQP (template, silvery white) (Fig. (S4a)). The two structures possessed high similarity, with an RMSD (root–mean–square deviation) of 0.77 Å. The model was composed of the VH and VL, which was in accord with typical features of antibodies. The secondary structure of the model is composed of 21 α–helices, 88 extended strands, 21 β–turns, and 100 random coil regions (Fig. (S4b)).
The quality of the model structure was first evaluated by inspecting its backbone conformation with the aid of the Ramachandran plot obtained from PROCHECK [Citation24]. The results are showed in . Ninety percent of the residues are in the core region, 9.5% residues in the allowed regions and only 0.5% in the disallowed regions (). These residues with an unfavorable backbone conformation were mainly located in the linker region of the model, which had no proper crystal structure as a homology model template. Verify_3D was also used to further validate the model. In this study, the 3D–1D score was used to verify the quality of the model. A 3D–1D score above 0.2 is considered to be a high–quality model, according to the rulers [Citation25]. The result of Verify_3D indicated that the averaged 3D–1D score for 89.97% residues was greater than 0.2. The structural analyses confirm the quality of the predicted model structure.
Figure 4. Validation of the homology model by Ramachandran analysis.
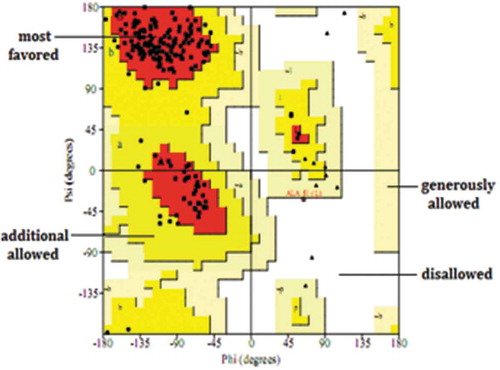
Figure 5. The variable regions of antibody interaction with PFLX. (a) The variable region 3D structure in complex with the ligand PFLX. (b) The pocket of the binding site. Residues that contribute most to the binding energy are represented as sticks model and labeled.
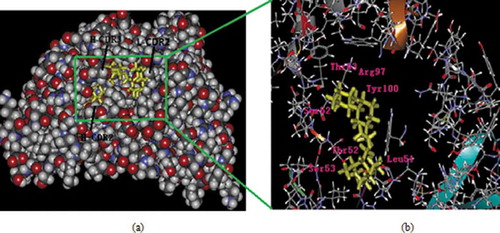
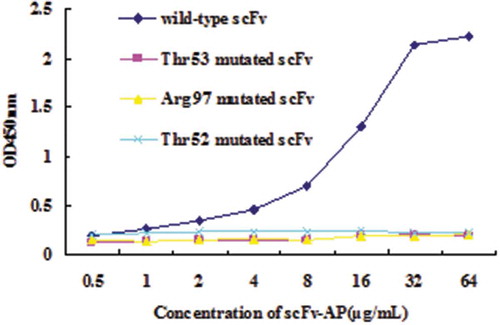
Figure 6. ELISA assay for mutated scFvs.
Docking of variable regions
To illustrate the interactive nature of the antibody and PFLX, the ligand-receptor docking of PFLX and the antibody was studied by DS software. Ninety–nine binding conformations were obtained. For the antibody–PFLX complex, the lowest binding energy (a measure of structural stability) was found to be ΔE = – 3.40 kcal/mol, according to the procedure to calculate the energy [Citation26].
The optimal structure of the receptor and the docked PFLX molecule is shown in ) and the docking site is magnified in ). The pocket of the active site adopts a groove, which is a hydrophobic environment. The PFLX moiety was buried in the pocket with the hydrophobic environment promotion. The active pocket is primarily formed by Ser52, Thr53, Arg97, Tyr100, Leu51, Thr52, and Ser53 residues. Ser52 and Thr53 residues belong to the VH–CDR2; Arg97 and Tyr100 belong to the VH–CDR3; Leu51, Thr52, and Ser53 belong to the VL–FR2. Three strength bonds are critical during antibody-ligand interaction. At the C–terminus, the piperazine ring were embedded into the receptor. The O2 and N1 atoms of PFLX formed H–bonds with an OH of Arg97 and NH2 of Thr53, respectively. Arg97 has a long flexible side chain, and this flexibility likely influences the affinity of the antibody; On the piperazine ring terminus, the N3 atom of PFLX formed H–bonds with an NH2 of Thr52, which dragged light chain deviated C-terminus. Besides these residues, van der Waals forces and surface electrostatic charges contribute a minor role to recognition.
Point mutation of scFv and validation docking
The point mutation scFv were induced with E. coli BL21, and the expressed products were analyzed with SDS–PAGE. As shown in Fig. S5, mutated scFv proteins (Lane 1, Lane 2 and Lane 3) were highly expressed.
After mutated scFv proteins were purified using affinity chromatography, ELISA was performed. As shown in , the wild-type scFv showed the highest binding activity, while the binding activity of the three mutated scFv has decreased greatly, showing a very low binding activity to PFLX antigen. The above result demonstrated that Thr53, Arg97 and Thr52 of scFv were critical for the interaction between antibody and PFLX antigen.
Discussion
A mAb specific for PFLX has been produced and a dc-ELISA was developed and successfully used to determine PFLX in milk samples. The assay was capable of determining PFLX in milk samples at concentrations as low as 0.15 µg/L. The method described here could be a promising analytical tool for sensitive determination of PFLX.
As to molecular recognition, a possible structure of the ligand-binding pocket and variable region of the antibody has been derived, and the docking results were verfied using point mutation. Three predicted essential amino acids, Thr53, Arg97 of heavy chain and Thr52 of light chain were critical for the interaction between antibody and PFLX antigen.
This is the first report on the interaction mechanism of PFLX and its antibody using homology modeling. The approach used in here can improve immunoassay features by facilitating the redesign of the immunizing hapten in animal tissue and also modify antibodies using orthomutation. The potential advantages are as follows: (i) Polyclonal and monoclonal antibodies of quinolone. Here, redesigning the immunizing and coating hapten to obtain antibodies with broader specificity and higher sensitivity. (ii) Modify the antibody using orthomutations. Genetically engineering an antibody as a new type of antibody has merits, including a low immunogenic antigen, cost effectiveness, easy to modify, and a potential commercial process. Therefore, the development of antibodies using this approach has potential. However, its affinity is so poor that it is not suitable to use in a clinical setting. The reasons for these potential drawbacks arise from nucleotide mutations in CDR regions and incorrect folding during production in cells. Protein modifications are the best method to improve this mAb. Here, we can directly mutate key amino acids to improve affinity.
Authors’ contributions
KH and XYZ conceived the approach, DHZ,XYZ and ZHH performed experiments. KH and XYZ wrote the manuscript. All authors read and approved the fnal manuscript.
Supplymentary_file.doc
Download MS Word (245.5 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Sun WY, Liu WY, Qu LB. Development of ELISA and immunochromatographic assay for ofloxacin. Chin Chem Lett. 2007;18:1107–1110.
- Zhao T, Yu BF, Tang WJ, et al. Spectrofluorimetric determination of ofloxacin in milk with N-(9-fluorenylmethyloxycarbonyl)-l-alanine. Spectrochim Acta. 2015;148:125–129.
- Hopkala H, Kowalczuk D. Application of derivative UV spectrophotometry for the determination of ciprofloxacin, norfloxacin and ofloxacin in tablets. Acta Poloniae Pharmaceutica. 2000;57:3–13.
- Basci NE, Hanioglu-Kargi S, Soysal H. Determination of ofloxacin in human aqueous humour by high performance liquid chromatography with fluorescence detection. J Pharm Biomed Anal. 1997;15:663–666.
- Horstkötter C, Blaschke G. Stereoselective determination of ofloxacin and its metabolites in human urine by capillary electrophoresis using laser-induced fluorescence detection. J Chromatogr B. 2001;754:169–178.
- Ev LDS, Schapoval EES. Microbiological assay for determination of ofloxacin injection. J Pharm Biomed Anal. 2002;27:91–96.
- Yuan M, Liu B, Liu EM, et al. Immunoassay for phenylurea herbicides: applicationof molecular modeling and quantitative structure activity relationship analysis on an antigen-antibody interaction study. Anal Chem. 2011;83:4767–4774.
- Zang S, Liu YJ, Lin MH. A dual amplified electrochemical immunosensor for ofloxacin: polypyrrolefilm-Au nanocluster as the matrix and multi-enzyme-antibody functionalized gold nanorod as the label. Electrochim Acta. 2013;90:246–253.
- Wang ZH, Zhang HY, Ni HJ, et al. Development of a highly sensitive and specific immunoassay for enrofloxacin based on heterologous coating haptens. Anal Chim Acta. 2014;820:152–158.
- Liang SZ, Sun XMWei D, et al. Production of multiclone antibody of plex. Hubei Agricultural Sciences. 2011;50:1222–1225.
- Liu YZ, Zhao GX, Wang P, et al. Production of the broad specific monoclonal antibody against sarafloxacin for rapid immunoscreening of 12 fluoroquinolones in meat. Environ Occup Health. 2013;48:139–146.
- Hu K, Huang XY, Jiang YS, et al. Monoclonal antibody based enzyme-linked immunosorbent assay for the specific detection of ciprofloxacin and enrofloxacin residues in fishery products. Aquaculture. 2010;310:8–12.
- Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 1987;196:901–917.
- Morea V, Lesk AM, Tramontano A. Antibody modeling: implications for engineering and design. Methods. 2000;20:267–279.
- Wang RZ, Huang AL, Liu LC, et al. Construction of a single chain variable fragment antibody (scFv) against tetrodotoxin (TTX) and its interaction with TTX. Toxicon Toxicon. 2014;83:22–34.
- Jiang JQ, Zhang HT, Qi YH. Production and characterization of monoclonal antibodies against norfloxacin. Procedia Environ Sci. 2011;8:529–535.
- Hou XL, Guo KJ, Bao J, et al. Development of an enzyme-linked immunosorbent assay for detection of ofloxacin residue in environment matrix. J Food Agric Environ. 2011;9:779–783.
- Dong JX, Li ZF, Lei HT, et al. Development of a single-chain variable fragment-alkaline phosphatase fusion protein and a sensitive direct competitive chemiluminescent enzymeimmunoassay for detection of ractopamine in pork. Anal Chim Acta. 2012;736:85–91.
- Xu ZL, Dong JX, Wang H, et al. Production and characterization of a single-chain variable fragment linked alkaline phosphatase fusion protein for detection of O,O-diethyl organophosphorus pesticides in a one-step enzyme-linked immunosorbent assay. J Agri Food Chem. 2012;60:5076–5083.
- Sheng W, Li YZ, Xu X, et al. Enzyme-linked immunosorbent assay and colloidal gold-based immunochromatographic assay for several (fluoro)quinolones in milk. Microchim Acta. 2011;173:307–316.
- Li YL, Ji BQ, Chen W, et al. Production of new class-specific polyclonal antibody for determination of fluoroquinolones antibiotics by indirect competitive ELISA. Food Agric Immunol. 2008;19:251–264.
- Wang ZH, Zhu Y, Ding SY, et al. Development of a monoclonal antibody-based broad-specificity ELISA for fluoroquinolone antibiotics in foods and molecular modeling studies of cross-reactive compounds. Anal Chem. 2007;79:4471–4483.
- Huet AC, Charlier C, Tittlemier SA, et al. Simultaneous determination of (Fluoro)quinolone antibiotics in kidney, marine products, eggs, and muscle by enzyme-linked immunosorbent assay (ELISA). J Agri Food Chem. 2006;54:2822–2827.
- Kiruakaran P, Muthusamy K, Singh KD, et al. Homology modeling, molecular dynamics, and molecular docking studies of Trichomonas vaginalis carbamate kinase. Med Chem Res. 2012;21:2105–2116.
- Jain CK, Gupta M, Prasad Y, et al. Homology modeling and protein engineering of alkane monooxygenase in Burkholderia thailandensis MSMB121: in silico insights. J Mol Model. 2014;20:2045–2049.
- Stefano DL, Alessandro A. Structural and functional insights on folate receptor α(FRα) by homology modeling, ligand docking and molecular dynamics. J Mol Graphics Modell. 2013;44:197–207.