
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
A concise synthesis of litseaones A and B, which were isolated from the stem barks of Litsea rubescens and L. pedunculata, is described in this study. Litseaone A was synthesized in just three steps from a known phloroglucinol derivative. The direct conversion of litseaone A into litseaone B was also achieved.
Graphical Abstract
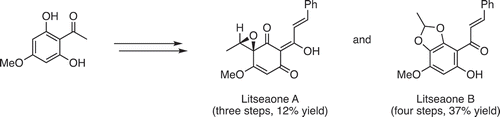
The concise synthesis of (±)-litseaones A and B was accomplished.
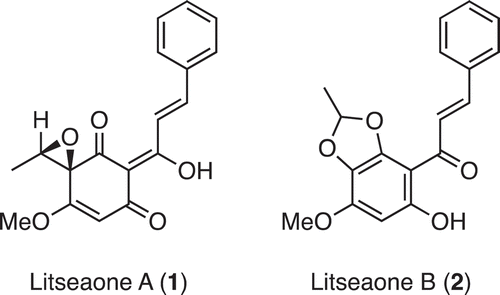
In 2011, Zhang et al. isolated litseaones A (1) and B (2) from the stem barks of Litsea rubescens and L. pedunculata [Citation1], which have been traditionally used for treating gastroenteralgia, edema and rheumatic arthritis [Citation2] (). Litseaones A (1) and B (2) have been reported to exhibit moderate cytotoxic activities against myeloid leukemia (HL-60) and epidermoid carcinoma (A431) cell lines. Recently, we reported the first asymmetric synthesis and structural revision of 1 [Citation3]. The key asymmetric reduction using (Ipc)2BCl enabled the synthesis of both enantiomers of 1. However, the use of a multistep protection/deprotection process made the synthesis rather cumbersome (nine steps), which inspired us to improve the synthetic efficiency. Herein, we describe a concise and protecting-group-free synthesis of (±)-1 and 2.
Figure 1. Structures of litseaones A (1) and B (2).
Result and discussion
shows our synthetic strategy toward litseaones A (1) and B (2). The conversion of 1 into 2 would be possible via the rearrangement reaction of the spiroepoxyketone. Compound 1 would be synthesized from 3 via oxidative dearomatization and subsequent aldol condensation with benzaldehyde. Intermediate 3 would be obtained from a known phloroglucinol derivative 4 [Citation4] via α-hydroxyethylation with acetaldehyde.
Scheme 1. Synthetic strategy for litseaones.
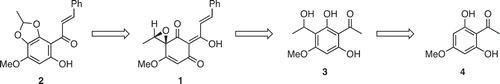
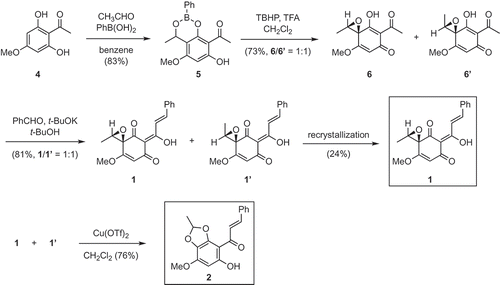
illustrates the synthesis of (±)-1 and 2. The known phloroglucinol derivative 4 was chosen as a starting material. The 3-(α-hydroxyethylation) of 4 with acetaldehyde in the presence of benzeneboronic acid [Citation5] afforded boronic ester 5 in 83% yield. Hydrolysis (or its equivalent operation) of the boronic ester function in 5 was then examined. However, several attempted conditions (KHF2, CH3CN/H2O; NaHCO3, THF/H2O; H2O2, NaOH, THF/H2O; pinacol, benzene) did not yield the desired product 3. After considerable experimentation, it was found that the treatment of 5 with t-butyl hydroperoxide [Citation6] and trifluoroacetic acid in CH2Cl2 directly produced an inseparable mixture of diastereomers 6 and 6ʹ in 73% yield (6/6ʹ = 1:1). The mixture of 6 and 6ʹ was then subjected to aldol condensation with benzaldehyde to give a 1:1 mixture of 1 and its diastereomer 1ʹ in 81% yield. Recrystallization from hexane/ethyl acetate produced pure litseaone A (1) in 24% yield. On the other hand, litseaone B (2) was obtained from a mixture of 1 and 1ʹ via Cu(OTf)2-mediated rearrangement of the spiroepoxyketone [Citation7] in 76% yield. 1H and 13C NMR spectra of our synthetic 1 and 2 were in good accordance with those of the natural compounds.
Scheme 2. Synthesis of (±)-1 and 2.
In conclusion, we accomplished the concise and protecting-group-free synthesis of (±)-1 (three steps and 12% overall yield) and (±)-2 (four steps and 37% overall yield) from the known phloroglucinol derivative 4. The synthesis described here is much more efficient than that previously reported for (+)-1 (nine steps and 1.7% overall yield) and (–)-1 (nine steps and 0.9% overall yield).
Experimental section
Dry THF was distilled from sodium-benzophenone ketyl and dry CH2Cl2 was distilled from P2O5 before use. Melting points are uncorrected values. IR spectra were measured on a JASCO FT/IR-230 spectrophotometer. 1H NMR was recorded on JEOL JNM ECS-400. Chemical shifts (δ) were reported to the residual solvent of CDCl3 (δ 7.26). 13C NMR was recorded on JEOL JNM ECS-400. Chemical shifts (δ) were reported relative to CDCl3 (δ 77.16). Mass spectra were recorded on JEOL JMS-T100LC. Column chromatography was performed on Kanto silica gel 60N (0.060–0.200 mm). TLC was carried out on Merck glass plates precoated with silica gel 60 F254 (0.25 mm).
1-(7-Hydroxy-5-methoxy-4-methyl-2-phenyl-4H-benzo[d][1,3,2]dioxaborinin-8-yl)ethan-1-one (5)
To a solution of 4 (370 mg, 2.03 mmol) and acetaldehyde (1.0 mL, 18 mmol) in benzene (20 mL) was added benzeneboronic acid (744 mg, 6.10 mmol) at room temperature under argon atmosphere. After stirring at room temperature for 22 h, the reaction mixture was concentrated in vacuo. Purification by silica gel column chromatography (eluted with hexane/EtOAc = 4:1 to 2:1) afforded 5 (530 mg, 83%) as a colorless solid: Mp 129–130°C; IR (KBr) νmax = 1627, 1590, 1370, 1311, 1248, 1122, 842, 693 cm−1; 1H NMR (400 MHz, CDCl3) δ = 1.54 (3H, d, J = 6.0 Hz), 2.93 (3H, s), 3.85 (3H, s), 5.37 (1H, q, J = 6.0 Hz), 6.20 (1H, s), 7.44 (2H, t, J = 7.2 Hz), 7.53 (1H, t, J = 7.2 Hz), 7.95 (2H, d, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ = 24.96, 33.59, 55.97, 65.83, 94.87, 105.90, 107.39, 128.00, 131.85, 134.43, 150.84, 161.14, 165.91, 203.25; ESI/TOFMS m/z calcd for C17H17BNaO5+ [M+Na]+ 335.1061, found 335.1038.
(2R,3S
)-5-Acetyl-8-methoxy-2-methyl-4-hydroxy-1-oxaspiro[2.5]octa-4,7-dien-6-one (6) and (2S
,3S
)-5-Acetyl-8-methoxy-2-methyl-4-hydroxy-1-oxaspiro[2.5]octa-4,7-dien-6-one (6ʹ)
To a solution of 5 (295 mg, 0.945 mmol) and t-butyl hydroperoxide (5–6M in decane solution, 2.0 mL, 10–12 mmol) in dry CH2Cl2 (20 mL) was added trifluoroacetic acid (217 µL, 2.84 mmol). After stirring for 1.5 h, phosphate buffer solution (pH = 6.8) was added to the mixture and the aqueous layer was extracted with EtOAc. The combined organic layer was dried over Na2SO4 and concentrated in vacuo. Purification by silica gel column chromatography (eluted with hexane/EtOAc = 8:1 to 4:1) afforded a mixture of 6 and 6ʹ (154 mg, 73%, dr = 1:1) as a yellow solid: IR (KBr) νmax = 1622, 1241, 1157, 1103, 990, 891, 846, 563 cm−1; 1H NMR (400 MHz, CDCl3) δ = 1.48 (1.5H, d, J = 5.2 Hz), 1.54 (1.5H, d, J = 5.2 Hz), 2.60 (3H, s), 3.43 (0.5H, q, J = 5.2 Hz), 3.63 (0.5H, q, J = 5.2 Hz), 3.84 (1.5H, s), 3.88 (1.5H, s), 5.66 (0.5H, s), 5.73 (0.5H, s); 13C NMR (100 MHz, CDCl3): δ = 11.30, 13.28, 28.07, 28.19, 56.94, 57.08, 60.83, 60.96, 63.97, 67.83, 98.95, 100.27, 107.87, 109.01, 170.22, 170.32, 185.94, 186.77, 188.43, 188.51, 200.77, 201.20; ESI/TOFMS m/z calcd for C11H12NaO5+ [M+Na]+ 224.0577, found 224.0563.
(2R,3R
,5E,2ʹE)-5-(1-Hydroxy-3-phenylallylidene)-8-methoxy-2-methyl-1-oxaspiro[2.5]oct-7-ene-4,6-dione (1)
To a solution of 6 and 6ʹ (48.5 mg, 0.217 mmol) and benzaldehyde (221 µL, 2.17 mmol) in t-butyl alcohol (4 mL) was added potassium t-butoxide (72.9 mg, 0.649 mmol) at room temperature under argon atmosphere. The reaction mixture was warmed to 70°C and stirred for 7 h. Saturated aqueous NH4Cl solution was added and the mixture was extracted with EtOAc. The combined organic layer was washed with water and brine, dried over Na2SO4, and concentrated in vacuo. Purification by silica gel column chromatography (eluted with hexane/EtOAc = 4:1 to 2:1) afforded a mixture of 1 and 1ʹ (54.4 mg, 81%, dr = 1:1) as a yellow solid. The mixture was recrystallized from hexane-EtOAc to afford 1 (16.1 mg, 24%) as yellow crystals: Mp 181–184°C {Ref [Citation1]. (+)-1: Mp 199–201°C}; IR (KBr) νmax = 1671, 1625, 1578, 1524, 1456, 1240, 1125 cm−1; 1H NMR (400 MHz, CDCl3) δ = 1.55 (3H, d, J = 5.6 Hz), 3.47 (1H, q, J = 5.6 Hz), 3.88 (3H, s), 5.80 (1H, s), 7.36–7.44 (3H, m), 7.62–7.71 (2H, m), 7.95 (1H, d, J = 15.0 Hz), 8.32 (1H, d, J = 15.0 Hz), 18.82 (1H, s); 13C NMR (100 MHz, CDCl3) δ = 13.43, 56.88, 61.30, 67.29, 102.29, 107.42, 122.63, 129.09, 129.15, 131.04, 135.14, 146.00, 169.69, 187.10, 187.40, 190.94; ESI/TOFMS m/z calcd for C18H16NaO5+ [M+Na]+ 335.0890, found 335.0897.
(E)-1-(5-Hydroxy-7-methoxy-2-methylbenzo[d] [1,3]dioxol-4-yl)-3-phenylprop-2-en-1-one (2)
To a solution of 1 and 1ʹ (14.8 mg, 0.0474 mmol) in dry CH2Cl2 (1 mL) was added Cu(OTf)2 (34.3 mg, 0.0948 mmol) at room temperature under argon atmosphere. After stirring for 70 min, water was added and the mixture was extracted with CH2Cl2. The combined organic layer was dried over Na2SO4 and concentrated in vacuo. Purification by silica gel column chromatography (eluted with hexane/EtOAc = 8:1) afforded 2 (11.2 mg, 76%) as a red solid: Mp 153-154ºC {Ref [Citation1]. (+)-2: Mp 158–160°C}; IR (KBr) νmax = 3063, 2853, 1659, 1593, 1515, 1443, 1385, 1345, 1291, 1231, 1164, 940, 871, 737 cm−1; 1H NMR (400 MHz, CDCl3) δ = 1.82 (3H, d, J = 4.8 Hz), 3.92 (3H, s), 6.13 (1H, s), 6.39 (1H, q, J = 4.8 Hz), 7.40–7.45 (3H, m), 7.59–7.65 (2H, m), 7.83 (1H, d, J = 15.6 Hz), 7.90 (1H, d, J = 15.6 Hz); 13C NMR (100 MHz, CDCl3) δ = 20.73, 56.47, 94.13, 102.16, 110.95, 124.71, 128.42, 128.84, 129.09, 130.76, 135.07, 144.45, 148.72, 150.20, 160.92, 190.42; ESI/TOFMS m/z calcd for C18H16NaO5+ [M+Na]+ 335.0890, found 335.0912.
Author Contribution
H.W. designed this study. H.K. carried out the experiments. H.T. contributed to analytical works. H.K. and N.M. wrote the manuscript with assistance from all authors.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Li L, Zhao X, Luo Y, et al. Novel cytotoxic chalcones from Litsea rubescens and Litsea pedunculata. Bioorg Med Chem Lett. 2011;21:7431–7433.
- Yang JH, Li L, Wang YS, et al. Two new aporphine alkaloids from Litsea glutinosa. Helv Chim Acta. 2005;88:2523–2526.
- Kageji H, Mori N, Takikawa H, et al. Synthesis and structure revision of litseaone A. Tetrahedron. 2018;74:7074–7081.
- Basabe P, Román MD, Marcos IS, et al. Prenylflavonoids and prenyl/alkyl-phloroacetophenones: synthesis and antitumour biological evaluation. Eur J Med Chem. 2010;45:4258–4269.
- Nagata W, Okada K, Aoki T. ortho-Specific α-hydroxyalkylation of phenols with aldehydes. An efficient synthesis of saligenol derivatives. Synthesis. 1979;1979(5):365–368.
- Píšová M, Souček M. Oxidative rearrangement of o-fuchsone to 2,2-diphenyl-1,3-benzodioxol. Collect Czech Chem Commun. 1982;47:3318–3327.
- Miller LA, Marsini MA, Pettus TRR. Chemoselective reactions of 3-benzyloxy-1,2-o-quinone with organometallic reagents. Org Lett. 2009;11:1955–1958.