
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
We conducted liquid chromatography-mass spectrometry measurements on hot-water extracts of peel from different varieties of Allium cepa. Some quercetin glycosides were identified as potential α-glucosidase inhibitors by principal component analysis of the liquid chromatography-mass spectrometry data. α-Glucosidase inhibitory activity assays identified quercetin-4ʹ-O-glucoside as an α-glucosidase inhibitor.
There are approximately 425 million diabetic patients worldwide, and this has led to growth in medical spending and associated social issues [Citation1]. The enzyme α-glucosidase (αG) breaks the α-1,4 bond of disaccharides and produces monosaccharides, which are absorbed into the body. Inhibition of this enzyme could prevent absorption of excess sugar, and inhibitors could be utilized as anti-diabetic medicines. Currently, some αG inhibitors like acarbose are used as medicines. In the food science field, research on αG inhibitors is popular and many inhibitors derived from foods have been reported [Citation2–Citation4].
Principal component analysis (PCA) is a multivariate analysis method that can be used for comprehensive data processing. This method is often applied to data from instrumental methods such as liquid chromatography/mass spectrometry (LC/MS), gas chromatography/mass spectrometry, and nuclear magnetic resonance spectroscopy, and has been utilized for determining producing areas of crops and evaluating deterioration of food products [Citation5–Citation9]. PCA makes it possible to detect slight differences among chromatograms. If samples are grouped according to differences in their function, it would be possible that we could pick up functional components easily from PCA data. Therefore, in this report, we used PCA to identify αG inhibitors contained in peel from different varieties of Allium cepa. First, we conducted LC/MS on hot-water extracts of peel from eight A. cepa varieties. We identified αG inhibitor quercetin-4ʹ-O-glucoside by PCA, instrumental analysis and comparison with standards.
Peel samples from the eight A. cepa varieties (onion, fresh onion, shallot, small onion, red onion, garlic, rakkyo brown, and rakkyo white) were freeze-dried, ground into fine powders, and extracted with hot water (10 mg of peel, 1 mL of water, 80°C, 30 min). The αG inhibition rates of the extracts were examined using a αG inhibition test. First, an αG enzyme solution was prepared as follows: intestinal acetone powder from rats (2 g) was added to 20 mL of phosphate buffer (pH 6.9, 0.1 M), centrifuged and the supernatant was retained and diluted five times with the buffer. For each sample, three wells were prepared in a 96-well plate. Each well contained 25 µL of the sample and 20 µL of enzyme solution. After shaking the plates, 25 µL of a substrate solution (4-nitrophenyl-α-d-glucopyranoside, 10 mM in pH 6.8 phosphate buffer) was added to each well. The enzyme reaction proceeded for 20 min at 37°C, and then 50 μL of 1 M Tris solution was added to stop the reaction. Blank wells were prepared containing the sample solution (sample blanks) or distilled water (control blanks), but enzyme solution was not added until after the Tris solution, which meant that no enzymatic reaction occurred. Control wells were prepared by adding 25 µL of distilled water to each well instead of the sample extract. The absorbance values at 405 nm were measured using a plate reader (µQuant, Biotek, Winooski, VT, USA). The inhibition rate for each extract was calculated using the following Equation (1)
where A is the absorbance in the sample well, A’ is the absorbance in the sample blank well, B is the absorbance in the control well, and B’ is the absorbance in the control blank well.
Hot-water extracts were filtered through three-layer filters (Whatman syringe filter, ø 25 mm, 0.45 µm) and injected into a LC-MS system (1260 Infinity binary LC and 6430 triple quadrupole LC-MS; Agilent Technologies Inc., California, USA). We followed the method described by Kammerer et al. with small modifications [Citation10]. We used the following components and parameters: column, Synergi Hydro-RP 100 A (100 mm × 3 mm, ø 2.5 µm; Phenomenex Inc., California, USA); column oven temperature, 40°C; mobile phase, 2% acetic acid (A) and 0.5% acetic acid/acetonitrile (1:1 v/v, B); mobile phase flow rate, 0.4 mL/min; injection volume, 5 µL; ion source, electrospray ionization (positive and negative modes); drying gas, nitrogen (350°C, 12 L/min); nebulizing gas, nitrogen (0.41 MPa); capillary voltage, ± 2500 V; fragmentor voltage, 100 V; and mass range, m/z 100–1000. A gradient elution was performed as follows: 0–8 min, 10%–24% B; 8–16 min, 24%–30% B; 16–24 min, 30%–55% B; 24–30 min, 55%–100% B; held at 100% until 33.2 min; and decreased to 10% and held at this level until 36 min. Product ion scan was conducted using the same system above. The accurate mass analysis was conducted for the same samples using LC-QTOF-MS system (LC20 A, Shimadzu, Kyoto, Japan and micrOTOF-Q II, Bruker, Massachusetts, USA). The LC conditions including columns were the same as the above. We used the following MS parameters; ion source, electrospray ionization (negative ion mode); mass range, 50–300; dry gass, nitrogen (200 °C, 8.0 L/min),; nebulizing gas, nitrogen (1.6 bar) and capillary voltage, 2800 V. Sodium acetate was used as an inner calibrant.
PCA was performed using the statistical software Pirouette® (Infometrix Inc., Washington, USA). Base peak chromatograms of samples transformed into ANDI format were aligned using the software LineUp™ (Infometrix Inc.). A data matrix of retention times and peak intensities was generated using PiroTrans ver. 1.35 (GL Science Inc., Tokyo, Japan). The peak intensities over 0.01 min intervals were averaged and normalized.
Among the eight A. cepa varieties, five (onion, small onion, shallot, fresh onion and red onion) showed relatively strong αG inhibition. PCA of these base peak chromatograms (positive and negative ion mode) yielded clusters of samples that did and did not inhibit αG ()). Peaks at around 13.1, 21.8, and 26.5 min were observed on the negative side of PC1 ()), which indicated the compounds causing these peaks contributed to αG inhibition since the cluster of samples that inhibited αG was observed on the negative side of PC1 of the score plot ()).
Figure 1. Principal component analysis score plot. (a) and loading plot. (b) of hot water extracts of peel from eight varieties of Allium cepa.
Each point in the score plot is for a sample and each point in the loading plot is for a retention time. In the loading plot, some characteristic points are marked.
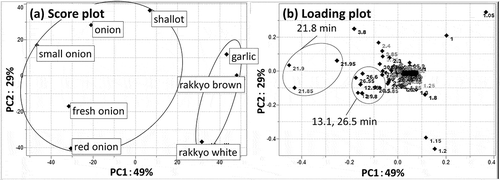
Accurate mass analysis gave molecular formulae of [M-H]− for the compounds responsible for the 13.1 (compound A), 21.8 (compound B), and 26.5 (compound C) min peaks of C27H30O17 (error: −0.5 ppm), C21H19O12 (error: −1.1 ppm), and C15H9O7 (error: −0.4 ppm), respectively. Product ion scan toward compounds above revealed that they had the same fragment ion (m/z 303, positive). In addition to that, fragment ions of m/z 627, 465 were detected for compound A, m/z 465 for compound B. The difference among each fragment ion is 162, which is equal to the molar weight of dehydrated hexoses. Results above indicated that compounds A and B were respectively diglycoside and monoglycoside of compound C. One of main components in onion peel is reportedly quercetin, and we selected quercetin (C15H10O7) and its mono and diglycoside (C21H20O12, C27H31O17) as candidate molecules for these peaks [Citation11].
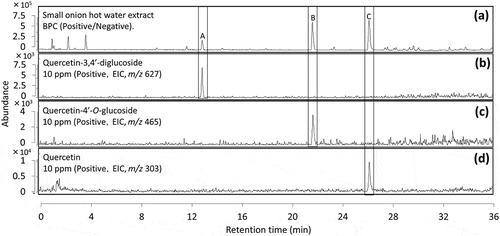
Standards of quercetin was purchased from Tokyo Chemical Industry (Tokyo, Japan), quercetin-4ʹ-O-glucoside (Q4G) and quercetin-3,4ʹ-diglucoside (QDG) from Funakoshi (Tokyo, Japan), quercetin-3-glucoside (Q3G) and quercetin-3-galactoside (Q3GA) from ChromaDex, (California, USA).We conducted comparison with standards, and identical retention times were observed for combinations of compound A and QDG (), compound B and Q4G (), compound C and quercetin (). From the above, we elucidated quercetin, Q4G and QDG as potential αG inhibitors. Besides, it is confirmed that Q3G and Q3GA were detected at different retention times compared to Q4G, which means that in this LC condition, it is possible to separate glycosides according to the difference in the binding sites and variety of sugars (data not shown).
Figure 2. LC/MS base peak chromatograms of a hot water extract of. (a)small onion and standards of. (b)quercetin-3,4ʹ-diglucoside, (c)quercetin-4ʹ-O-glucoside, and (d)quercetin.
Peaks at the retention time of 13.1, 21.8 and 26.5 min are marked as A, B and C respectively.
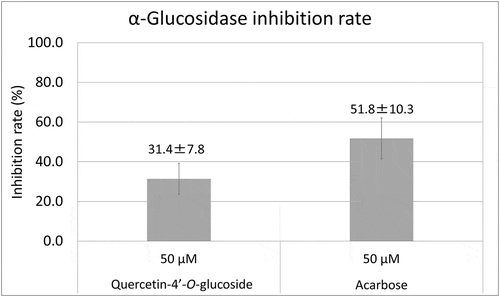
Among these three compounds, quercetin is already known for its αG inhibition. Therefore we conducted αG inhibition assays for the other two compounds and acarbose as a positive control [Citation12]. Since no inhibition was observed for 50 µM QDG (4.3 ± 5.0%, average±SD), we compared the rest of two and Q4G showed inhibition close to acarbose (). It is reported the main driving force of the interaction between quercetin and αG is hydrophobic [Citation12]. This could explain the difference of inhibition rates between Q4G and QDG. As the amount of sugar increases, the molecule becomes less hydrophobic, which connects to weaker interaction with αG. This could be one of reasons why Q4G showed stronger inhibition toward αG than the diglycoside. In summary, we tried to identify αG inhibitors in peel of eight A. cepa varieties using LC/MS and PCA. We extracted and identified quercetin-4ʹ-O-glucoside as a novel αG inhibitor.
Figure 3. α-Glucosidase inhibition rates of quercetin-4ʹ-O-glucoside and acarbose (n = 3, average±SD).
For each standard, inhibition rates are for 50μM solutions. No significant difference was observed by Welch’s t test. Acarbose was used as a positive control.
Author Contribution
Conceived and designed the experiments: Hidenobu Sumitani. Performed the experiments and the PCA: Kayako Ogi. Contributed to the writing of the manuscript: Kayako Ogi and Hidenobu Sumitani. All the authors approved the final version of the manuscript.
Acknowledgments
We thank Gabrielle David from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Cho NH, Shaw JE, Karuranga S, et al. IDF diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. 2018;138:271–281.
- Yu Z, Yin Y, Zhao W, et al. Novel peptides derived from egg white protein inhibiting alpha-glucosidase. Food Chem. 2011;129:1376–1382.
- Tadera K, Minami Y, Takamatsu K, et al. Inhibition of α-glucosidase and α-amylase by flavonoids. J Nutr Sci Vitaminol. 2006;52:149–153.
- Li DQ, Qian ZM, Li SP. Inhibition of three selected beverages extracts on α-glucosidase and rapid identification of their active compounds using HPLC-DAD-MS/MS and biochemical detection. J Agric Food Chem. 2010;58:6608–6613.
- Kim J, Jung Y, Bong YS, et al. Determination of the geographical origin of kimchi by 1H NMR-based metabolite profiling. Biosci Biotechnol Biochem. 2012;76(9):1752–1757.
- Mehari B, Redi-Abshiro M, Singh Chandravanshi B, et al. Characterization of the cultivation region of Ethiopian coffee by elemental analysis. Anal Lett. 2016;49:2474–2489.
- Ordouri SA, Cagliani LR, Lalou S, et al. 1H-NMR-based metabolomics of saffron reveals markers for its quality deterioration. Food Res Int. 2015;70:1–6.
- Upadhyay R, Mishra HN. Mutivariate analysis for kinetic modeling of oxidative stability and shelf life estimation of sunflower oil blended with sage (Salvia officinalis) extract under rancimat conditions. Food Bioprocess Technol. 2015;8(4):801–810.
- Ogi K, Otsuka T, Sumitani H. Isomerization of feruloylputrescine in orange juice by light exposure. Biosci Biotechnol Biochem. 2018;82(1):127–134.
- Kammerer D, Claus A, Carle R, et al. Polyphenol screening of pomace from red and white grape varieties (Vitis vinifera L.) by HPLC-DAD-MS/MS. J Agric Food Chem. 2004;252:4360–4367.
- Rohdes MJC, Price KR. Analytical problems in the study of flavonoid compounds in onions. Food Chem. 1996;57:113–117.
- Li YQ, Zhou FC, Gao F, et al. Comparative evaluation of quercetin, isoquercetin and rutin as inhibitors of α-glucosidase. J Agric Food Chem. 2009;57:11463–11468.