
ABSTRACT
Excessive uric acid production, which causes gout and hyperuricemia, can be blocked by inhibiting xanthine oxidase (XO). However, some agents to block on XO often cause side effects, thereby necessitating the identification of new inhibitors. During the screening of XO inhibitors from various mushroom extracts, we found that a methanolic extract of the fruiting bodies of Tyromyces fissilis, an inedible and non-toxic fungus, showed inhibitory activity. Both n-hexane and ethyl acetate layers, obtained by partitioning this extract exhibited XO inhibitory activity. Subsequently, using an activity-guided separation method, eight active compounds (1–8) were isolated. The structures of five of the new compounds, 2–4, 6, and 7, were elucidated by spectral analysis and chemical derivatization. All compounds had a salicylic acid moiety with an aliphatic group at the C-6 position. Notably, 2-hydroxy-6-pentadecylbenzoic acid (1) showed the highest level of XO noncompetitive inhibition (58.9 ± 2.2% at 25 µM).
Graphical abstract
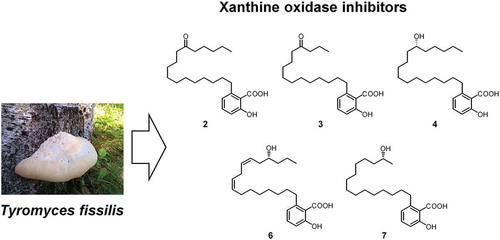
Compounds 2–4, 6, and 7 newly isolated from the fruiting bodies of Tyromyces fissilis.
Uric acid, the causative agent of gout, is the final product of purine (a nucleic acid) metabolism, which is excreted in urine. It is produced by the successive oxidation of hypoxanthine and xanthine by the catalytic enzyme xanthine oxidase (XO) [Citation1]. When excess uric acid accumulates in blood, the urate crystals tend to precipitate and aggregate at relatively low temperatures. A painful inflammation occurs when white blood cells attack the deposited crystals, leading to the symptoms collectively known as gout [Citation2]. The onset of gout is determined by the plasma uric acid level. When this level reaches 7.0 mg/dL or more, the patient is diagnosed with hyperuricemia [Citation3], which has been correlated with cardiovascular diseases [Citation4], hypertension [Citation5], and renal diseases [Citation6]. To treat both gout and hyperuricemia, the XO inhibitors, allopurinol and febuxostat, which inhibit uric acid production, were developed [Citation7]. However, treatment using these inhibitors has been associated with various side-effects, such as the development of hepatic disorders. Therefore, the discovery of new uric acid production inhibitors that have a low risk of causing side-effects is desired [Citation8] and has led to increased interest in natural products, particularly XO-inhibiting phytochemicals, including flavonoids, phenylpropanoids, anacardic acids [Citation9], hispidin derivatives [Citation10], and berchemiosides [Citation11].
Tyromyces fissilis is a white-rot fungus of the Polyporaceae family that grows on fallen broad-leaved trees from summer to autumn [Citation12]. While the fruiting bodies are hard and unsuitable for consumption, various components have been isolated and tested for their activities. For example, several lanosterol derivatives, whose physiological activities have yet to be clarified [Citation13,Citation14], and 2-hydroxybenzoic acid analogues, which appear to have anti-inflammatory activities [Citation15], have been isolated from this species of fungus. Furthermore, a screening of mushroom extracts for XO enzymatic inhibition demonstrated that a methanolic extract of T. fissilis exhibited the highest inhibitory activity (data not shown).
In the present study, we analyzed possible XO inhibitors that were isolated from T. fissilis fruiting bodies by methanol extraction. To our knowledge, this is the first study that has conducted the isolation, structural determination, and inhibitory activity of the XO inhibitors derived from T. fissilis.
Materials and methods
Fungal material
Fruiting bodies of T. fissilis were collected and provided by Mr. Mizuki Nomura and Dr. Mitsuru Hirota of the Ina Campus, Shinshu University, Japan in October 2014. The fungal material was identified by Dr. Akiyoshi Yamada of the Faculty of Agriculture, Shinshu University. A voucher specimen (Mito-01) was deposited in a laboratory in the Faculty of Agriculture, Shinshu University.
Chemicals
XO (EC 1.17.3.2, from bovine milk), xanthine, 1-methyl-3-nitro-1-nitrosoguanidine, Dess-Martin periodinane, and 4-(dimethylamino) pyridine (DMAP) were obtained from Sigma Chemical Company. Ethylenediaminetetraacetic acid (EDTA) was purchased from Nacalai Tesque Inc. Deuterochloroform (CDCl3) was obtained from Merck. Methanol (MeOH), n-hexane, n-butanol (n-BuOH), acetonitrile (MeCN), chloroform (CHCl3), and ethyl acetate (EtOAc) were obtained from Kanto Chemical Company. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC∙HCl), (1R,2R)-2-(anthracene-2,3-dicarboximido)cyclohexanecarboxylic acid, and (1S,2S)-2-(anthracene-2,3-dicarboximido)cyclohexanecarboxylic acid were obtained from the Tokyo Chemical Industry Corporation.
General experimental methods
One and two dimentional nuclear magnetic resonance (NMR) spectra, including the 1H-1H correlation spectroscopy (COSY), heteronuclear multiple quantum correlation (HMQC), and heteronuclear multiple bond correlation (HMBC) spectra, were recorded on the Bruker DRX500 spectrometer operated at 500 MHz for 1H and 125 MHz for 13C. 1H chemical shifts were determined relative to the internal standard tetramethylsilane (TMS). 13C chemical shifts were determined using the CDCl3 solvent peak (δC 77.0) as the reference. High-resolution fast atom bombardment mass spectra (HR-FAB-MS) were obtained on the JEOL JMS-700MStation spectrometer with glycerol as the matrix. Optical rotations were measured using the JASCO DIP-100 digital polarimeter (L = 1 cm). Melting points were measured using a Yanaco MP-S3 measuring device. IR spectra were recorded on the JASCO FT/IR-480 Plus spectrometer, while UV spectra were measured on the Shimadzu UV-1800 spectrometer. The silica gels, silica gel 60 F254 and 60 RP-18 F254S (Merck), were used for analytical thin-layer chromatography (TLC). Column chromatography was performed using a normal-phase silica gel (Fuji Silysia BW-350) and reverse-phase octadecyl functionalized silica gel (ODS; Fuji Silysia BM1020T). High-performance liquid chromatography (HPLC) was performed on the Shimadzu system (Kyoto, Japan) consisting of the LC-10AD pump, CTO-10A column oven, and SPD-10A UV-VIS detector, which were connected to the C-R7A plus Chromatopac integrator. A reverse-phase column (10ϕ × 250 cm, 5 µm, Inertsil ODS-3), flow rate of 3 mL/min, and wavelength of 254 nm were used for the analysis. All solvents were HPLC-grade.
Isolation of compounds 1–8
Fruiting bodies of T. fissilis (2.1 kg) were soaked in MeOH (4 L) for 4 months. After filtration and evaporation of MeOH in vacuo, the resulting crude extract was dissolved in H2O (250 mL) and then successively partitioned with n-hexane (500 mL, thrice), EtOAc (500 mL, thrice), and n-BuOH (375 mL, thrice). The dried n-hexane extract (9.29 g), EtOAc extract (39.4 g), and n-BuOH extract (7.37 g) were then evaluated for XO inhibition. The n-BuOH extract showed low XO inhibition (4.5% at 100 µg/mL), while the n-hexane and EtOAc extracts showed relatively high inhibitory activity against XO with inhibitory rates of 28.5% and 37.8%, respectively, at 100 µg/mL (The XO inhibitory activities of these extracts are shown in Supplementary Data).
The n-hexane extract was further subjected to silica gel column chromatography. Elution was performed using a stepwise mixture of n-hexane/EtOAc (100:0, 90:10, 80:20, 70:30, and 0:100, v/v; 100 mL each) and MeOH, resulting in 27 fractions (TFH1–TFH27) based on TLC analysis. Fraction TFH4 (a part of the 90% n-hexane/EtOAc eluate, 302.5 mg) was subsequently subjected to additional silica gel column chromatography with a stepwise mixture of n-hexane/EtOAc (100:0, 99:1, 98:2, 97:3, 96:4, 95:5, 94:6, 93:7, 92:8, 91:9, and 90:10, v/v; 30 mL each) and MeOH as the eluent, resulting in 12 additional fractions (TFH4-1 to TFH4-12). The MeOH eluate, TFH4-12 (34.7 mg), was further separated using the ODS column with MeCN/H2O (90:10, 91:9, 92:8, and 93:7, v/v; 5 mL each) as the eluent to give compound 1 (19.7 mg). The mixture of the TFH14 and TFH15 fractions (parts of the 80% n-hexane/EtOAc eluate) (60.4 mg) was fractionated using the ODS column with MeCN/H2O (60:40, 65:35, 70:30, 75:25, and 80:20, v/v; 30 mL each) as the eluent, resulting in 15 subfractions [TFH(14,15)-1 to TFH(14,15)-15)]. The 80% MeCN/H2O eluate (subfractions 13–15, 17.5 mg) was mixed and separated by reverse-phase HPLC (80:20, v/v; MeCN/H2O) to give compound 2 (5.0 mg). Three parts of the 80% n-hexane/EtOAc eluate, TFH16–18 (58.8 mg), were subjected to ODS flash column elution with MeCN/H2O (60:40, 65:35, 70:30, 75:25, and 80:20, v/v; 15 mL each) as the eluent to give compound 3 (9.0 mg). Part of the 80% n-hexane/EtOAc eluate, TFH21 (32.8 mg), was similarly separated to obtain compound 4 (20.5 mg). Part of the 70% n-hexane/EtOAc eluate, TFH25 (152.4 mg), was fractionated using the ODS column with MeCN/H2O (60:40, 65:35, 70:30, 75:25, 80:20, and 85:15, v/v; 60 mL each) as the eluent to produce 11 subfractions (1–11). Part of the 75% MeCN/H2O eluate TFH25-6 (26.0 mg) was separated by recrystallization from MeCN to give compound 5 (12.9 mg).
2-Hydroxy-6-pentadecylbenzoic acid (1): Colorless amorphous powder; mp 86–89°C; UV (EtOH) λmax (log ε) 209 (4.47), 243 (3.77), and 310 (3.58) nm; IR (NaCl) νmax 3500–2500, 2916, 2853, 1657, 1605, 1448, 1308, 1249, 882, 815, 730, 708, 647, and 596 cm−1; HR-FAB-MS m/z 347.2584 [M−H]− (calcd. for C22H35O3, 347.2586); 1H NMR (500 MHz, CDCl3) δ 0.88 (3H, t, J = 7.1 Hz), 1.20–1.38 (24H, m), 1.57 (2H, m), 2.94 (2H, dd, J = 7.6, 7.9 Hz), 6.75 (1H, d, J = 7.5 Hz), 6.84 (1H, d, J = 8.2 Hz), and 7.31 (1H, dd, J = 7.5, 8.2 Hz); 13C NMR (125 MHz, CDCl3) δ 14.1, 22.7, 29.4, 29.5, 29.7, 29.7, 29.8, 31.9, 32.0, 36.3, 111.2, 115.7, 122.7, 135.0, 147.6, 163.0, and 176.1.
2-Hydroxy-6-(12-oxoheptadecyl)benzoic acid (2): Colorless amorphous powder; mp 93–94°C; UV (EtOH) λmax (log ε) 209 (4.48), 243 (3.79), and 311 (3.60) nm; IR (NaCl) νmax 3500–2500, 2916, 2850, 1705, 1652, 1607, 1449, 1299, 1222, 907, 813, and 720 cm−1; HR-FAB-MS m/z 389.2686 [M−H]− (calcd. for C24H37O4, 389.2692); 1H and 13C NMR, see and .
Table 1. 13C NMR spectral data for 2–4, 6, and 7 (125 MHz, CDCl3).
Table 2. 1H NMR spectral data for 2–4, 6, and 7 (500 MHz, CDCl3).
2-Hydroxy-6-(12-oxopentadecyl)benzoic acid (3): Colorless amorphous powder; mp 82–84°C; UV (EtOH) λmax (log ε) 209 (4.40), 243 (3.70), and 310 (3.50) nm; IR (NaCl) νmax 3500–2500, 2922, 2850, 1706, 1654, 1608, 1449, 1301, 1218, 903, 814, and 721 cm−1; HR-FAB-MS m/z 361.2377 [M−H]− (calcd. for C22H33O4, 361.2379); 1H and 13C NMR, see and .
(R)-2-Hydroxy-6-(12-hydroxyheptadecyl)benzoic acid (4): Colorless amorphous powder; mp 75–76°C; [α]18.7D −0.5° (c 0.42, MeOH); UV (EtOH) λmax (log ε) 209 (4.44), 243 (3.75), and 310 (3.55) nm; IR (NaCl) νmax 3500–2500, 3446, 2926, 2854, 1653, 1605, 1452, 1311, 1246, 1214, 1166, 824, and 709 cm−1; HR-FAB-MS m/z 391.2838 [M−H]− (calcd. for C24H39O4, 391.2848); 1H and 13C NMR, see and .
(R)-2-Hydroxy-6-(12-hydroxypentadecyl)benzoic acid (5): Colorless oil; [α]19.9D −1.0° (c 0.19, MeOH); UV (EtOH) λmax (log ε) 209 (4.41), 243 (3.71), and 310 (3.52) nm; IR (NaCl) νmax 3500–2500, 3451, 2925, 2853, 1647, 1605, 1451, 1310, 1246, 1213, 1166, 823, and 742 cm−1; HR-FAB-MS m/z 363.2546 [M−H]− (calcd. for C22H35O4, 363.2535); 1H NMR (500 MHz, CDCl3) δ 0.94 (3H, t, J = 6.6 Hz), 1.23–1.40 (14H, m), 1.40–1.54 (8H, m), 1.58 (2H, m), 2.96 (2H, m), 3.70 (1H, m), 6.75 (1H, d, J = 7.5 Hz), 6.85 (1H, d, J = 8.3 Hz), and 7.33 (1H, dd, J = 7.5, 8.3 Hz); 13C NMR (125 MHz, CDCl3) δ 14.1, 18.8, 25.2, 28.7, 28.9, 28.9, 29.0, 29.0, 29.1, 29.6, 32.1, 36.5, 37.0, 39.5, 72.5, 110.9, 115.6, 122.5, 134.9, 147.5, 163.5, and 174.6.
The EtOAc extract was subjected to silica gel column chromatography with the n-hexane/EtOAc mixture (100:0, 90:10, 80:20, 70:30, 60:40, 50:50, 40:60, 30:70, 20:80, 10:90, 0:100, v/v; 400 mL each) and MeOH as the eluent, producing 13 fractions (TFE1–TFE13) based on solvent composition and TLC analyses. Part of the 70% n-hexane/EtOAc eluate, TFE4 (2764.1 mg), was further subjected to silica gel column chromatography with an n-hexane/EtOAc mixture (100:0, 90:10, 80:20, 70:30, 60:40, 50:50, and 40:60, v/v; 240 mL each) as the eluent, affording 25 subfractions (TFE4-1 to TFE4-25). Two parts of the 70–80% n-hexane/EtOAc eluate, TFE4-9 and TFE4-10 (599.1 mg), were mixed and dissolved in MeCN and then, the supernatant (178.3 mg) was further fractionated using the ODS column with MeCN/H2O (60:40, 65:45, 70:30, 75:25, 80:20, and 85:15, v/v; 70 mL each) as the eluent to give 36 subfractions (1–36). Of these, subfractions 6 and 7 (14.8 mg), which are parts of the 70–75% MeCN/H2O eluate, were subjected to silica gel column chromatography with a CHCl3/MeOH (100:0, 98:2, and 96:4, v/v; 4.8 mL each) as the eluent to obtain compound 6 (7.7 mg). Part of the 60% n-hexane/EtOAc eluate, TFE5 (393.2 mg), was subjected to additional silica gel column chromatography with an n-hexane/EtOAc mixture (100:0, 90:10, 80:20, 70:30, 60:40, and 50:50, v/v; 30 mL each) as the eluent, resulting in 22 additional fractions (TFE5-1 to TFE5-22). Part of the 70% n-hexane/EtOAc eluate, TFE5-11 (111.2 mg), was further fractionated using the ODS column with MeCN/H2O (60:40, 65:35, 70:30, 75:25, and 80:20, v/v; 45 mL each) as the eluent to afford 22 subfractions (1–22). Two parts of the 75% MeCN/H2O eluate, subfractions 7 and 8 (25.4 mg) were further separated by recrystallization from MeCN to give compound 7 (20.1 mg). The MeCN eluate, TFE5-11–22 (5.3 mg), was similarly separated and recrystallized from MeCN to give compound 8 (5.2 mg).
2-Hydroxy-6-((R,8Z,11Z)-14-hydroxyheptadeca-8,11-dien-1-yl)benzoic acid (6): Colorless oil; [α]18.7D +2.6° (c 0.17, methanol); UV (EtOH) λmax (log ε) 204 (4.39), 240 (3.62), and 309 (3.39) nm; IR (NaCl) νmax 3500–2500, 3436, 3008, 2926, 2854, 1653, 1605, 1451, 1308, 1247, 1214, 1166, 1123, 1014, 823, and 708 cm−1; HR-FAB-MS m/z 387.2538 [M−H]− (calcd. for C24H35O4, 387.2535); 1H and 13C NMR, see and .
(R)-2-Hydroxy-6-(14-hydroxypentadecyl)benzoic acid (7): Colorless amorphous powder; mp 74–76°C; [α]18.7D −3.8° (c 0.40, methanol); UV (EtOH) λmax (log ε) 209 (4.43), 243 (3.74), and 311 (3.55) nm; IR (NaCl) νmax 3500–2500, 3451, 2925, 2853, 1653, 1605, 1451, 1310, 1246, 1214, 1166, 823, and 708 cm−1; HR-FAB-MS m/z 363.2540 [M−H]− (calcd. for C22H35O4, 363.2535); 1H and 13C NMR, see and .
(R)-2-Hydroxy-6-(16-hydroxyheptadecyl)benzoic acid (8): Colorless amorphous powder; mp 83–84°C; [α]22.0D −4.2° (c 0.11, methanol); UV (EtOH) λmax (log ε) 209 (4.36), 243 (3.66), and 310 (3.47) nm; IR (NaCl) νmax 3500–2500, 3470, 2916, 2849, 1683, 1575, 1470, 1314, 1248, 1104, 823, 772, 716, and 701 cm−1; HR-FAB-MS m/z 391.2858 [M−H]− (calcd. for C24H39O4, 391.2848); 1H NMR (500 MHz, CDCl3) δ 1.22 (3H, d, J = 6.2 Hz), 1.24–1.55 (26H, m), 1.59 (2H, m), 2.96 (2H, dd, J = 7.1, 8.7 Hz), 3.87 (1H, m), 6.76 (1H, d, J = 7.5 Hz), 6.86 (1H, d, J = 8.3 Hz), and 7.34 (1H, dd, J = 7.5, 8.3 Hz); 13C NMR (125 MHz, CDCl3) δ 23.3, 25.6, 29.2, 29.3, 29.3, 29.4, 29.4, 29.5, 29.9, 32.1, 36.5, 39.1, 68.7, 110.7, 115.7, 122.6, 135.0, 147.5, 163.5, and 174.7.
Methylation of compounds 1, 2, and 4–8 by diazomethane
The method used to obtain the methyl esters of 1, 2, and 4–8 was based on that described by Nomura et al. [Citation15]. For example, compound 1 (1.44 mg) was dissolved in MeOH (3 mL). An excess amount of diazomethane (in Et2O) was then added, and the reaction mixture was stirred at room temperature for 15 h. Subsequently, methyl ester 9 (1.33 mg) was obtained from 1 by removing the solvents in vacuo from the reaction mixture. Compounds 2, and 4–8 (2.00, 4.68, 4.55, 0.54, 5.56, and 1.20 mg, respectively) were also treated similarly to obtain methyl ester derivatives 10 (2.98mg), 11 (5.21mg), 12 (4.62mg), 13 (0.45mg), 14 (6.16mg), and 15 (1.48 mg), respectively.
Methyl 2-methoxy-6-pentadecylbenzoate (9): Colorless amorphous powder; 1H NMR (500 MHz, CDCl3) δ 0.88 (3H, t, J = 7.0 Hz), 1.20–1.35 (24H, m), 1.52–1.62 (2H, m), 2.53 (2H, dd, J = 7.8, 8.0 Hz), 3.82 (3H, s), 3.91 (3H, s), 6.76 (1H, d, J = 8.3 Hz), 6.82 (1H, d, J = 7.7 Hz), and 7.27 (1H, dd, J = 7.7, 8.3 Hz).
Methyl 2-methoxy-6-(12-oxoheptadecyl)benzoate (10): Colorless amorphous powder; 1H NMR (500 MHz, CDCl3) δ 0.89 (3H, t, J = 7.3 Hz), 1.20–1.34 (18H, m), 1.56 (6H, m), 2.38 (4H, t, J = 7.5 Hz), 2.53 (2H, dd, J = 7.7, 8.1 Hz), 3.82 (3H, s), 3.91 (3H, s), 6.76 (1H, d, J = 8.3 Hz), 6.82 (1H, d, J = 7.7 Hz), and 7.27 (1H, dd, J = 7.7, 8.3 Hz); 13C NMR (125 MHz, CDCl3) δ 13.9, 22.5, 23.6, 23.9, 29.3, 29.4, 29.5, 29.5, 29.6, 31.2, 31.4, 33.5, 42.8, 42.8, 52.2, 55.8, 108.3, 121.5, 123.4, 130.2, 141.3, 156.2, 169.0, and 211.8.
Methyl (R)-2-(12-hydroxyheptadecyl)-6-methoxybenzoate (11): Colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.89 (3H, t, J = 6.9 Hz), 1.21–1.36 (18H, m), 1.35–1.49 (8H, m), 1.53–1.61 (2H, m), 2.53 (2H, dd, J = 7.8, 8.1 Hz), 3.55–3.62 (1H, m), 3.82 (3H, s), 3.91 (3H, s), 6.76 (1H, d, J = 8.3 Hz), 6.82 (1H, d, J = 7.7 Hz), and 7.27 (1H, dd, J = 7.7, 8.3 Hz).
Methyl (R)-2-(12-hydroxypentadecyl)-6-methoxybenzoate (12): Colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.93 (3H, t, J = 6.7 Hz), 1.20–1.36 (14H, m), 1.36–1.50 (8H, m), 1.52–1.61 (2H, m), 2.53 (2H, dd, J = 7.8, 8.0 Hz), 3.57–3.64 (1H, m), 3.82 (3H, s), 3.91 (3H, s), 6.76 (1H, d, J = 8.3 Hz), 6.82 (1H, d, J = 7.7 Hz), and 7.27 (1H, dd, J = 7.7, 8.3 Hz).
Methyl 2-((R,8Z,11Z)-14-hydroxyheptadeca-8,11-dien-1-yl)-6-methoxybenzoate (13): Colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.93 (3H, t, J = 6.4 Hz), 1.20–1.40 (10H, m), 1.40–1.52 (2H, m), 1.52–1.62 (2H, m), 2.04 (2H, q, J = 7.0 Hz), 2.24 (2H, t, J = 6.7 Hz), 2.53 (2H, dd, J = 7.8, 8.0 Hz), 2.81 (2H, t, J = 6.8 Hz), 3.62–3.68 (1H, m), 3.82 (3H, s), 3.91 (3H, s), 5.29–5.35 (1H, m), 5.36–5.42 (1H, m), 5.44 (1H, dt, J = 7.5, 10.6 Hz), 5.55 (1H, dt, J = 7.3, 10.7 Hz), 6.76 (1H, d, J = 8.3 Hz), 6.82 (1H, d, J = 7.7 Hz), and 7.27 (1H, dd, J = 7.7, 8.3 Hz).
Methyl (R)-2-(14-hydroxypentadecyl)-6-methoxybenzoate (14): Colorless amorphous powder; 1H NMR (500 MHz, CDCl3) δ 1.18 (3H, d, J = 6.2 Hz), 1.23–1.35 (20H, m), 1.35–1.50 (2H, m), 1.57 (2H, m), 2.53 (2H, dd, J = 7.8, 8.0 Hz), 3.79 (1H, m), 3.82 (3H, s), 3.91 (3H, s), 6.76 (1H, d, J = 8.3 Hz), 6.82 (1H, d, J = 7.7 Hz), and 7.27 (1H, dd, J = 7.7, 8.3 Hz).
Methyl (R)-2-(16-hydroxyheptadecyl)-6-methoxybenzoate (15): Colorless amorphous powder; 1H NMR (500 MHz, CDCl3) δ 1.19 (3H, d, J = 6.2 Hz), 1.22–1.35 (24H, m), 1.35–1.50 (2H, m), 1.52–1.62 (2H, m), 2.53 (2H, dd, J = 7.8, 8.0 Hz), 3.79 (1H, m), 3.82 (3H, s), 3.91 (3H, s), 6.76 (1H, d, J = 8.3 Hz), 6.82 (1H, d, J = 7.7 Hz), and 7.27 (1H, dd, J = 7.7, 8.3 Hz).
Oxidation of methylated derivative 11 by Dess-Martin periodinane
Compound 11 was oxidized via the method described by Nomura et al. [Citation15]. In CH2Cl2 (2 mL) of 11 (2.61 mg), an excessive amount of Dess-Martin periodinane was added. The resulting solution was stirred at room temperature for 2 h and extracted twice with EtOAc (5.0 mL). The combined EtOAc extracts were then evaporated to dryness and fractionated by silica gel column chromatography with n-hexane/EtOAc (80:20, v/v) as the eluent to obtain oxidation derivative 16 (2.41 mg).
Methyl 2-methoxy-6-(12-oxoheptadecyl)benzoate (16): Colorless amorphous powder; HR-FAB-MS m/z 417.2979 [M−H]− (calcd. for C26H41O4, 417.3005); 1H NMR (500 MHz, CDCl3) δ 0.89 (3H, t, J = 7.1 Hz), 1.20–1.36 (18H, m), 1.56 (6H, m), 2.38 (4H, t, J = 7.5 Hz), 2.53 (2H, dd, J = 7.8, 8.0 Hz), 3.82 (3H, s), 3.91 (3H, s), 6.76 (1H, d, J = 8.3 Hz), 6.82 (1H, d, J = 7.7 Hz), and 7.27 (1H, dd, J = 7.7, 8.3 Hz); 13C NMR (125 MHz, CDCl3) δ 13.9, 22.5, 23.6, 23.9, 29.3, 29.4, 29.5, 29.5, 29.6, 31.1, 31.4, 33.5, 42.8, 42.8, 52.2, 55.8, 108.3, 121.5, 123.4, 130.2, 141.3, 156.2, 169.0, and 211.8.
Esterification of compounds 11–15 with (1R,2R)- or (1S,2S)-reagent
The method described by Ohtaki et al. [Citation16] was used for the esterification of derivatives 11–15. (1R,2R)-2-(Anthracene-2,3-dicarboximido)cyclohexanecarboxylic acid (1.2 eq.) was dissolved in a mixture of toluene and acetonitrile (1:1,v/v; 200 µL). Derivatives 11–15 (1.0, 1.0, 0.2, 0.8, and 0.4 mg, respectively) were added to the solution along with EDC∙HCl (10 eq.) and DMAP (10 mg). The mixture was stirred overnight at room temperature, and then evaporated in vacuo. The mixture was purified by silica gel column chromatography to give the corresponding (1R,2R)-esters, 17 (0.98 mg), 19 (2.1 mg), 21 (0.1 mg), 23 (0.84 mg), and 25 (0.24 mg). In the same manner, derivatives 11–15 (1.0, 1.0, 0.2, 0.49, and 0.4 mg, respectively) were esterified with (1S,2S)-2-(anthracene-2,3-dicarboximido)cyclohexanecarboxylic acid and then purified by chromatography to afford the corresponding (1S,2S)-esters, 18 (0.23 mg), 20 (0.34 mg), 22 (0.1 mg), 24 (0.51 mg), and 26 (0.17 mg).
(R: 1R,2R)-Ester (17) formed from derivative 11: Yellow powder; 1H NMR (500 MHz, CDCl3) δ 0.70 (3H, t, J = 6.9 Hz), 0.91–0.95 (8H, m), 0.91–1.03 (5H, m), 1.03–1.13 (6H, m), 1.13–1.23 (3H, m), 1.23–1.35 (5H, m), 1.42–1.61 (4H, m), 1.85–1.91 (3H, m), 2.15–2.25 (2H, m), 2.51 (2H, dd, J = 7.8, 8.0 Hz), 3.56 (1H, dt, J = 3.4, 11.8 Hz), 3.82 (3H, s), 3.91 (3H, s), 4.49 (1H, dt, J = 3.6, 11.9 Hz), 4.74 (1H, m), 6.77 (1H, d, J = 8.3 Hz), 6.82 (1H, d, J = 7.7 Hz), 7.28 (1H, dd, J = 7.7, 8.3 Hz), 7.60 (2H, m), 8.06 (2H, m), 8.47 (2H, s), and 8.61 (2H, s).
(R: 1S,2S)-Ester (18) formed from derivative 11: Yellow powder; 1H NMR (500 MHz, CDCl3) δ 0.55 (3H, t, J = 7.0 Hz), 0.85–1.09 (12H, m), 1.09–1.18 (3H, m), 1.18–1.35 (12H, m), 1.35–1.66 (4H, m), 1.81–1.92 (3H, m), 2.13–2.23 (2H, m), 2.52 (2H, dd, J = 7.6, 8.1 Hz), 3.56 (1H, dt, J = 3.2, 12.4 Hz), 3.82 (3H, s), 3.91 (3H, s), 4.49 (1H, dt, J = 3.4, 12.2 Hz), 4.73 (1H, m), 6.76 (1H, d, J = 8.4 Hz), 6.82 (1H, d, J = 7.6 Hz), 7.29 (1H, dd, J = 7.6, 8.4 Hz), 7.61 (2H, m), 8.07 (2H, m), 8.48 (2H, s), and 8.62 (2H, s).
(R: 1R,2R)-Ester (19) formed from derivative 12: Yellow oil; 1H NMR (500 MHz, CDCl3) δ 0.71 (3H, t, J = 7.3 Hz), 0.75–0.90 (8H, m), 0.90–1.03 (4H, m), 1.03–1.23 (8H, m), 1.23–1.38 (3H, m), 1.38–1.63 (4H, m), 1.83–1.93 (3H, m), 2.13–2.27 (2H, m), 2.51 (2H, dd, J = 6.0, 7.8 Hz), 3.56 (1H, dt, J = 3.5, 11.8 Hz), 3.82 (3H, s), 3.91 (3H, s), 4.49 (1H, dt, J = 3.7, 12.0 Hz), 4.75 (1H, m), 6.77 (1H, d, J = 8.3 Hz), 6.82 (1H, d, J = 7.7 Hz), 7.29 (1H, dd, J = 7.7, 8.3 Hz), 7.60 (2H, m), 8.06 (2H, m), 8.48 (2H, s), and 8.61 (2H, s).
(R: 1S,2S)-Ester (20) formed from derivative 12: Yellow oil; 1H NMR (500 MHz, CDCl3) δ 0.51 (3H, t, J = 7.3 Hz), 0.85–1.08 (10H, m), 1.08–1.20 (3H, m), 1.20–1.35 (10H, m), 1.40–1.65 (4H, m), 1.81–1.93 (3H, m), 2.13–2.25 (2H, m), 2.52 (2H, dd, J = 7.5, 8.2 Hz), 3.57 (1H, dt, J = 3.8, 11.5 Hz), 3.82 (3H, s), 3.91 (3H, s), 4.49 (1H, dt, J = 3.5, 12.3 Hz), 4.75 (1H, m), 6.76 (1H, d, J = 8.2 Hz), 6.82 (1H, d, J = 7.7 Hz), 7.29 (1H, dd, J = 7.7, 8.2 Hz), 7.61 (2H, m), 8.08 (2H, m), 8.48 (2H, s), and 8.63 (2H, s).
(R: 1R,2R)-Ester (21) formed from derivative 13: Yellow oil; 1H NMR (500 MHz, CDCl3) δ 0.69 (3H, t, J = 7.3 Hz), 0.83–0.98 (2H, m), 1.08–1.35 (13H, m), 1.35–1.69 (4H, m), 1.79–1.93 (5H, m), 1.93–2.28 (4H, m), 2.47 (2H, m), 2.53 (2H, dd, J = 7.2, 8.4 Hz), 3.55 (1H, m), 3.81 (3H, s), 3.90 (3H, s), 4.47 (1H, dt, J = 4.8, 11.3 Hz), 4.77 (1H, t, J = 6.4 Hz), 4.97–5.12 (3H, m), 5.12–5.20 (1H, m), 6.76 (1H, d, J = 8.5 Hz), 6.82 (1H, d, J = 8.0 Hz), 7.23–7.30 (1H, m, overlapped with CHCl3), 7.62 (2H, m), 8.07 (2H, m), 8.48 (2H, s), and 8.62 (2H, s).
(R: 1S,2S)-Ester (22) formed from derivative 13: Yellow oil; 1H NMR (500 MHz, CDCl3) δ 0.45 (3H, t, J = 7.2 Hz), 0.80–1.08 (5H, m), 1.08–1.40 (10H, m), 1.38–1.70 (4H, m), 1.80–1.97 (5H, m), 2.12–2.23 (4H, m), 2.53 (2H, dd, J = 7.9, 8.0 Hz), 3.51 (1H, m), 3.81 (3H, s), 3.90 (3H, s), 4.47 (1H, m), 4.76 (1H, m), 5.12–5.23 (2H, m), 5.23–5.32 (2H, m), 6.75 (1H, d, J = 8.8 Hz), 6.81 (1H, d, J = 7.6 Hz), 7.23–7.30 (1H, m, overlapped with CHCl3), 7.62 (2H, m), 8.09 (2H, m), 8.48 (2H, s), and 8.63 (2H, s).
(R: 1R,2R)-Ester (23) formed from derivative 14: Yellow powder; 1H NMR (500 MHz, CDCl3) δ 0.80–0.90 (6H, m), 0.90–0.98 (2H, m), 1.00 (3H, d, J = 6.2 Hz), 1.05–1.13 (2H, m), 1.13–1.20 (4H, m), 1.20–1.35 (6H, m), 1.35–1.65 (4H, m), 1.80–1.93 (3H, m), 2.10–2.30 (2H, m), 2.54 (2H, dd, J = 7.8, 8.0 Hz), 3.50 (1H, dt, J = 3.8, 8.3 Hz), 3.82 (3H, s), 3.91 (3H, s), 4.47 (1H, dt, J = 3.8, 13.5 Hz), 4.74 (1H, m), 6.76 (1H, d, J = 8.2 Hz), 6.83 (1H, d, J = 7.7 Hz), 7.27 (1H, dd, J = 7.7, 8.2 Hz), 7.61 (2H, m), 8.07 (2H, m), 8.48 (2H, s), and 8.62 (2H, s).
(R: 1S,2S)-Ester (24) formed from derivative 14: Yellow powder; 1H NMR (500 MHz, CDCl3) δ 0.88 (3H, d, J = 6.2 Hz), 0.92–1.15 (12H, m), 1.15–1.35 (11H, m), 1.40–1.66 (4H, m), 1.80–1.93 (3H, m), 2.13–2.29 (2H, m), 2.53 (2H, dd, J = 7.8, 8.0 Hz), 3.52 (1H, dt, J = 3.1, 11.9 Hz), 3.82 (3H, s), 3.91 (3H, s), 4.44 (1H, dt, J = 3.5, 12.3 Hz), 4.74 (1H, m), 6.76 (1H, d, J = 8.3 Hz), 6.82 (1H, d, J = 7.7 Hz), 7.26–7.28 (1H, m, overlapped with CHCl3), 7.62 (2H, m), 8.08 (2H, m), 8.49 (2H, s), and 8.63 (2H, s).
(R: 1R,2R)-Ester (25) formed from derivative 15: Yellow powder; 1H NMR (500 MHz, CDCl3) δ 0.82–0.89 (10H, m), 0.89–0.98 (3H, m), 1.00 (3H, d, J = 6.2 Hz), 1.04–1.37 (13H, m), 1.37–1.72 (5H, m), 1.82–1.95 (3H, m), 2.10–2.27 (2H, m), 2.54 (2H, dd, J = 7.6, 8.1 Hz), 3.50 (1H, m), 3.82 (3H, s), 3.91 (3H, s), 4.46 (1H, m), 4.73 (1H, m), 6.76 (1H, d, J = 8.0 Hz), 6.82 (1H, d, J = 7.7 Hz), 7.25–7.30 (1H, m, overlapped with CHCl3), 7.61 (2H, m), 8.08 (2H, m), 8.48 (2H, s), and 8.63 (2H, s).
(R: 1S,2S)-Ester (26) formed from derivative 15: Yellow powder; 1H NMR (500 MHz, CDCl3) δ 0.83–0.92 (2H, m), 0.88 (3H, d, J = 6.1 Hz), 0.92–1.09 (10H, m), 1.09–1.36 (14H, m), 1.36–1.73 (5H, m), 1.80–1.93 (3H, m), 2.13–2.27 (2H, m), 2.53 (2H, dd, J = 8.0, 8.1 Hz), 3.52 (1H, m), 3.82 (3H, s), 3.91 (3H, s), 4.45 (1H, m), 4.75 (1H, m), 6.76 (1H, d, J = 8.6 Hz), 6.82 (1H, d, J = 7.9 Hz), 7.25–7.30 (1H, m, overlapped with CHCl3), 7.62 (2H, m), 8.08 (2H, m), 8.49 (2H, s), and 8.63 (2H, s).
XO inhibitory activity of the isolated compounds
The XO inhibitory activity was evaluated by measuring the uric acid level according to the method described by Masuoka et al. [Citation9]. The reaction mixture consisted of 2.76 mL of 40 mM sodium carbonate buffer containing 0.1 mM EDTA (pH 10.0), 0.06 mL of 10 mM xanthine, and 0.06 mL of the sample solution (dissolved in DMSO). The reaction was started by the adding 0.12 mL of XO solution (0.04 U), and the absorbance at 293 nm was recorded for 90 s. Control experiments were carried out by replacing the sample solution with the same amount of DMSO and/or by omitting xanthine in the reaction mixture. The XO inhibitory activity, in terms of the rate of uric acid production inhibition (%), was calculated as follows:
XO inhibition (%) = {1 – (Asample – Asample (no xanthine))/(Acontrol – Acontrol (no xanthine))} × 100
Results and discussion
Using the XO inhibitory activity-guided separation method described by Nguyen et al. [Citation17] (see Supplementary Data), we isolated 1–8 from the fruiting bodies of T. fissilis through methanol extraction, followed by various types of chromatographic separation (). Compounds 1, 5, and 8 were identified as 2-hydroxy-6-pentadecylbenzoic acid [Citation18], (R)-2-hydroxy-6-(12-hydroxypentadecyl)benzoic acid [Citation15], and (R)-2-hydroxy-6-(16-hydroxyheptadecyl)benzoic acid [Citation19], respectively, by comparing their physicochemical properties and spectral data to those in the literature.
Figure 1. Chemical structures of 1–8.
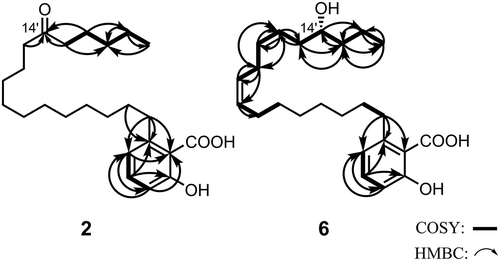
The negative HR-FAB-MS of 2 showed a deprotonated molecular ion at m/z 389.2686 [M−H] – (calcd. 389.2692), indicating that the molecular formula is C24H38O4 with five unsaturations. The IR spectrum suggested the presence of a carboxy (3500–2500 and 1652 cm−1), isolated carbonyl (1705 cm−1), and aromatic (907, 813, and 720 cm−1) groups. The 13C NMR spectrum indicates the presence of one carbonyl (δC 213.0), one carboxy (δC 175.0), one benzene (δC 110.6, 115.7, 122.6, 135.1, 147.6, and 163.5), one methyl (δC 13.9), and 15 methylene (δC 22.4, 23.6, 23.8, 29.0, 29.1, 29.2, 29.2, 29.2, 29.3, 29.6, 31.4, 32.0, 36.5, 42.8, and 42.8) groups (). The deduced substituted form of the aromatic ring is 1,2,3-trisubstituted benzene based on the coupling constants (J) of the aromatic proton signals at δH 6.77 (1H, d, J = 7.5 Hz), 6.86 (1H, d, J = 8.3 Hz), and 7.35 (1H, dd, J = 7.5, 8.3 Hz) in the 1H NMR spectrum (). These data are similar to those of 1 except for the signal owing to a ketone, and the aromatic group is presumed to be salicylic acid with an alkyl side chain at the C-6 position. This is confirmed by the presence of long-range correlations between H-1′ and C-1, C-5 and C-6, and H-2′ and C-6 in the HMBC spectrum.
The methylene protons adjacent to the ketone are observed at δH 2.41 (4H, t, J = 7.5 Hz) (, ). The correlation with the four methylene protons is observed in order from the terminal methyl proton δH 0.89 (3H, t, J = 7.2 Hz) to the fourth methylene proton at δH 2.41 in the 1H-1H COSY spectrum (), indicating that the length of the alkyl side chain is C17. The HMBC spectrum also exhibited C–H correlations between the three methylene protons (δH 2.41 and 1.57) and carbonyl carbon (δC 213.0). This indicates that the position of the carbonyl carbon is C-12′. Thus, 2 was determined as 2-hydroxy-6-(12-oxoheptadecyl)benzoic acid, which is a new compound.
Figure 2. Important COSY (bold) and HMBC (→) correlations for 2 and 6.
The NMR and IR profiles of 3 were similar to those of 2, indicating that 3 also has C6-substituted salicylic acid with alkyl side chain. The negative HR-FAB-MS confirmed that molecular formula of 3 is C22H34O4 (found: m/z 361.2377 [M−H]−). The 13C NMR spectrum also indicates the presence of 22 carbons, as shown in . These data directly show that 3 has a C15 alkyl side chain with one carbonyl group. The 1H-1H COSY spectrum suggested the presence of a partial structure based on the correlations between a terminal methyl proton (H-15′, δH 0.91) and methylene protons (H-14′, δH 1.60) and between two methylene protons (H-14′, δH 1.60 and H-13′, δH 2.40). Furthermore, the HMBC spectrum showed correlations between the carbonyl carbon (δC 212.9) and two methylene protons (H-13′, δH 2.40 and H-14′, δH 1.60). Thus, the carbonyl group was assigned to the C-12′ position in the alkyl side chain. These spectral data indicated that 3 is another new compound, specifically 2-hydroxy-6-(12-oxopentadecyl)benzoic acid.
The negative HR-FAB-MS confirmed that 4 has the molecular formula, C24H40O4 (found: m/z 391.2838 [M−H]–), indicating that its mass number is 2H more than that of 2. Its 13C NMR spectrum is similar to that of 2, suggesting the presence of the C6-substituted salicylic acid moiety with C15 alkyl side chain. The difference between 2 and 4 is the absence of the carbonyl signal at δC 213.0 for the latter; a new signal (δC 72.7) for oxymethine appears instead (). Based on detailed analysis of the HMQC and HMBC spectra, this oxymethine group is expected to be at C-12′ position in the alkyl side chain.
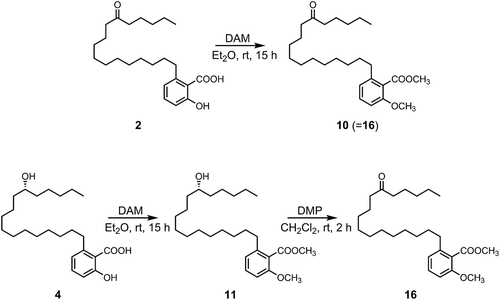
The methylation of 4 by diazomethane afforded methyl ester 11, which was then treated with Dess-Martin periodinane to oxidize the hydroxy group and, form derivative 16 (). The spectral data of 16 were completely identical to those of 10; comparison with the methylated 2 indicates that the hydroxy group of 4 is positioned at C-12′. Thus, compound 4 was identified as the new compound, 2-hydroxy-6-(12-hydroxyheptadecyl)benzoic acid.
Scheme 1. Chemical derivatization of 2 and 4.
The negative HR-FAB-MS of 6 showed a deprotonated molecular ion at m/z 387.2538 [M−H] – (calcd. 387.2535), indicating that the molecular formula is C24H36O4 with six unsaturations. The 13C NMR spectrum indicates the presence of one carboxy (δC 174.1), four olefinic (δC 124.7, 127.4, 130.6, and 132.0), one benzene (δC 111.0, 115.6, 122.4, 134.7, 147.3, and 163.5), one oxymethine (δC 72.0), one methyl (δC 14.0), and 11 methylene (δC 18.8, 25.8, 26.8, 28.5, 28.5, 29.0, 29.8, 32.2, 34.9, 36.7, and 38.4) groups (). The 1H NMR signals at δH 6.74 (1H, d, J = 7.5 Hz), 6.84 (1H, d, J = 8.2 Hz), and 7.32 (1H, dd, J = 7.5, 8.2 Hz) indicate the presence of a 1,2,3-trisubstituted benzene ring moiety (). Thus, 6 is also assumed to contain a derivative of the C6-substituted salicylic acid, which has a C17 side chain with C–C double bonds and an oxymethine group.
From the H–H correlations in the COSY spectrum was deduced the sequence from the terminal methyl group (δH 0.95) to the four olefinic protons, that is between methyl: H-17′ (δH 0.95), methylenes: H-16′ (δH 1.41) and H-15′ (δH 1.53), oxymethine: H-14′ (δH 3.81) and H-13′ (δH 2.33), olefinic proton: H-12′ (δH 5.43), olefinic proton: H-11′ (δH 5.60), olefinic proton: H-10′ (δH 2.83), olefinic proton: H-9 (δH 5.37), and olefinic proton: H-8 (δH 5.38), as shown in . Thus, the position of oxymethine and the two double bonds are C-14′, C-12′, C-11′, C-9′, and C-8′, respectively. The HMBC data also supported this assignment.
Based on the J values of the olefinic protons at δH 5.37 to 5.38 (11.0 Hz) and 5.43 to 5.60 (10.6 Hz), the configuration of the double bonds is Z. Compound 6 was determined as the new compound, 2-hydroxy-6-((8Z,11Z)-14-hydroxyheptadeca-8,11-dien-1-yl)benzoic acid.
Compound 7 has the same molecular formula, C22H36O4, as that of 5. Because its 1H and 13C NMR spectra are also similar to that of 5, it was suspected to be a positional isomer of 5 at the hydroxy group. Detailed assignment of the NMR signals based on the 1H-1H COSY and HMBC spectra indicated a hydroxy group at the C-14′. Thus, compound 7 was determined as 2-hydroxy-6-(14-hydroxypentadecyl)benzoic acid as a new compound.
Because compounds 4–8 have an asymmetric carbon in the alkyl side chain, their absolute configurations were determined using the method described by Ohrui et al. [Citation16,Citation20]. Compounds 4–8 have several overlapping signals in their 1H NMR spectra owing to the long alkyl side chain; thus, the data was difficult to analyze using the modified Mosher’s method as it requires many hydrogen assignments [Citation21]. Therefore, we used this method, whose empirical rule between the absolute configuration and chemical shift at the terminal methyl of the alkyl side chain of the secondary alcohol is known. First, the phenolic hydroxy and carboxy groups of 4–8 were methylated with diazomethane to produce the corresponding methyl esters, 9–15. Each methyl ester was then derivatized using (1R,2R)- or (1S,2S)-2-(anthracene-2,3-dicarboximido)cyclohexanecarboxylic acid to form the (1R,2R)- or (1S,2S)-ester, respectively. The (R)- and (S)- configurations of the asymmetric carbon atoms were judged by comparing the chemical shifts of the terminal methyl group of the alkyl side chain of the (1R,2R)- and (1S,2S)-esters for each compound.
The ester derivatives undergo a change in the 1,3-syn configuration of the carbonyl oxygen and the hydrogen on the α-carbon of the alcohol, as well as in the s-trans configuration of the carbonyl oxygen and the α-hydrogen at the C2 position of the cyclohexane moiety as shown in .
Figure 3. Structural representation of the chemical shifts observed for the terminal methyl group of the side chain.
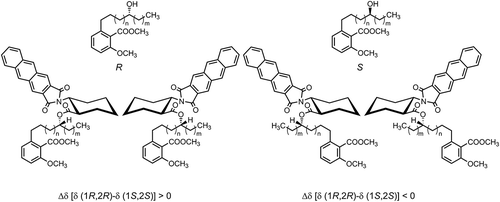
For the (R)-ester, the change in the chemical shift of the terminal methyl, {Δδ [δ (1R,2R) − δ (1S,2S)] ppm}, is positive, whereas that of the (S)-ester is negative. Therefore, because the differences between the chemical shifts of the (1R,2R)-esters and (1S,2S)-esters of 4–8 are positive (), their hydroxy groups have the (R)-configuration. Compounds 2–4, 6, and 7 are newly isolated compounds, while the (S)-isomers of 5 and 8 have not been reported thus far.
Table 3. Differences in the chemical shifts for the terminal methyl of the esters.
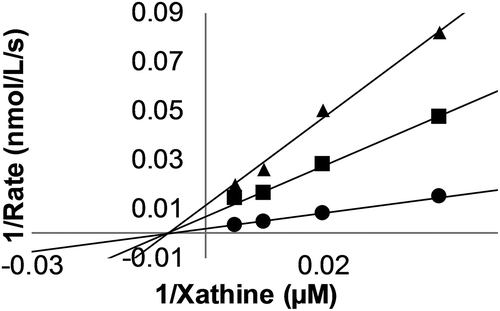
Finally, we evaluated the XO inhibitory activities of all isolated compounds and their derivatives following the method described by Masuoka et al. [Citation9] (). Allopurinol was chosen as the positive control. Among the tested compounds, compound 1 exhibits the highest level of XO inhibition at a concentration of 25 μM (inhibitory activity = 58.9 ± 2.2%), which is comparable to that of the positive control (inhibitory activity = 64.0 ± 0.6%). Furthermore, XO inhibition by 1 is noncompetitive, as shown in . Compound 6, which has two cis- double bonds in its alkyl side chain, shows the next highest level of inhibition, 30.6 ± 0.7% at a concentration of 100 μM. Interestingly, methylated derivative 9, exhibits a remarkably low activity compared with its parent compound (1), suggesting that the carboxy and hydroxy groups of the salicylic acid moiety are essential for XO inhibition. Although further investigation is necessary, the existence of the salicylic acid moiety and level of the side chain hydrophobicity seems to play a significant role on XO inhibitory activity, based on the correlation between the latter and the structure. This supports previous experimental results showing that the salicylic acid moiety of anacardic acid caused cooperative inhibition, while the alkenyl side chain enhanced this effect by interacting with the hydrophobic site of XO [Citation9].
Table 4. Xanthine oxidase inhibitory activity of 1–9 (n = 3).
Figure 4. Lineweaver-Burk plots of uric acid formation by XO. Multiple xanthine concentrations (0–50 μM) were oxidized by XO in the presence of varying concentrations of 1 (●, 0 µM; ■, 25 µM; and ▲, 50 µM). Each data point represents the average of three tests.
The compounds that have a salicylic acid moiety with an aliphatic group at the C-6 position are known as bioactive phytochemicals [Citation22]. For example, compound 1 has been shown to have inhibitory activity against glyceraldehyde-3-phosphate dehydrogenase [Citation18] and antitumor activity [Citation23]. The known compounds, 5 and 8, exhibit inflammation suppressive [Citation15] and tyrosinase inhibitory activities [Citation24]. Because a few anacardic acid derivatives isolated from edible plants, such as cashew nuts [Citation25], cashew apple [Citation26], mango peel [Citation27], the newly isolated compounds from T. fissilis are expected to show various biological activities, such as enzyme inhibition and antitumor activity, without causing a side effect.
Conclusions
In this study, eight XO-inhibiting compounds (1–8), including five new compounds (2–4, 6, and 7), were isolated from the fruiting bodies of T. fissilis, and their chemical structures were elucidated by instrumental analysis and chemical derivatization. In summary, these results indicated a significant structure-activity relationship, whereby the presence of a salicylic acid moiety and hydrophobicity of the alkyl side chain were found to be essential for XO inhibition. Although additional work is required to evaluate the biological activity of these compounds from fungus, this study provides a new framework for further investigation on the structure-related activity of XO inhibitors.
Author contributions
Conceived the work: T. F. and M. H. Designed the work: T. F. Isolation and structure determination of compounds: S. M. and T. F. Instrumental analysis and derivatization of compounds: S. M. Wrote the manuscript: S. M. and T. F. Reviewed the manuscript: M. H.
Supplementary_data_180809.docx
Download MS Word (3.3 MB)Acknowledgments
We are grateful to Dr. Yasuko Okamoto (Tokushima Bunri University) for performing the HR-FAB-MS measurements. We also thank Mr. M. Nomura and Prof. M. Hirota (Shinshu University) for collecting and providing the mushroom. This work was supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) KAKENHI under Grant Number JP15H01751.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Porras AG, Olson JS, Palmer G. The reaction of reduced xanthine oxidase with oxygen. Kinetics of peroxide and superoxide formation. J Biol Chem. 1981;256:9096–9103.
- Martinon F. Mechanisms of uric acid crystal-mediated autoinflammation. Immunol Rev. 2010;233:218–232.
- Kratz A, Pesce MA, Basner RC, et al. Laboratory values of clinical importance. In: Longo DL, Fauci AS, Kasper DL, et al., editors. Harrison’s principles of internal medicine. 18th ed. New York (NY): McGraw-Hill; 2011; p.1091–1095.
- Li M, Hu X, Fan Y, et al. Hyperuricemia and the risk for coronary heart disease morbidity and mortality a systematic review and dose-response meta-analysis. Sci Rep. 2016;6:19520.
- Ono I, Hosoya T. Chronic kidney diseases and various other diseases: 7. Hyperuricemia. Nihon Naika Gakkai Zasshi [Journal of the Japanese Society of Internal Medicine]. 2007;96:922–927. Japanese.
- Johnson RJ, Kang DH, Feig D, et al. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension. 2003;41:1183–1190.
- Borghi C, Desideri G. Urate-lowering drugs and prevention of cardiovascular disease: the emerging role of xanthine oxidase inhibition. Hypertension. 2016;67:496–498.
- Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450–2461.
- Masuoka N, Kubo I. Characterization of xanthine oxidase inhibition by anacardic acids. Biochim Biophys Acta. 2004;1688:245–249.
- Kemami Wangun HV, Hertweck C. Squarrosidine and Pinillidine: 3,3′-Fused bis(styrylpyrones) from Pholiota squarrosa and Phellinus pini. Eur. J Org Chem. 2007;2007:3292–3295.
- Kang KB, Park EJ, Kim J, et al. Berchemiosides A-C, 2-Acetoxy-ω-phenylpentaene fatty acid triglycosides from the unripe fruits of Berchemia berchemiaefolia. J Nat Prod. 2017;80:2778–2786.
- Imazeki R, Ohtani K, Hongou T. Nihon no kinoko [Japanese mushrooms]. Tokyo: Yama-kei Publishers; 1988. Japanese.
- Quang DN, Hashimoto T, Tanaka M, et al. Tyromycic acids B–E, new lanostane triterpenoids from the mushroom Tyromyces fissilis. J Nat Prod. 2004;67:148–151.
- Quang DN, Hashimoto T, Tanaka M, et al. Tyromycic acids F and G: two new triterpenoids from the mushroom Tyromyces fissilis. Chem Pharm Bull. (Tokyo). 2003;51:1441–1443.
- Nomura M, Hirota M Himematutake baiyou kinshi oyobi simitake sizitutai ni hukumareru ensyou yokusei busshitu [Inflammation suppressive substances contained in fruiting bodies of Agaricus subrufescens and Tyromyces fissilis] [master’s thesis]. Japan: Shinshu University; 2014. Japanese.
- Ohtaki T, Akasaka K, Kabuto C, et al. Chiral discrimination of secondary alcohols by both 1H-NMR and HPLC after labeling with a chiral derivatization reagent, 2-(2,3-anthracenedicarboximide)cyclohexane carboxylic acid. Chirality. 2005;17 Suppl:S171–S176.
- Nguyen MT, Awale S, Tezuka Y, et al. Xanthine oxidase inhibitory activity of Vietnamese medicinal plants. Biol Pharm Bull. 2004;27:1414–1421.
- Pereira JM, Severino RP, Vieira PC, et al. Anacardic acid derivatives as inhibitors of glyceraldehyde-3-phosphate dehydrogenase from Trypanosoma cruzi. Bioorg Med Chem. 2008;16:8889–8895.
- Shan R, Anke H, Nielsen M, et al. The isolation of two new fungal inhibitors of 35S-TBPS binding to the brain GABAA/benzodiazepine chloride channel receptor complex. Nat Prod Lett. 1994;4:171–178.
- Ohrui H. Development of highly potent chiral discrimination methods that solve the problems of diastereomer method. Proc Jpn Acad Ser B Phys Biol Sci. 2007;83:127–135.
- Ohtani I, Kusumi T, Kashman Y, et al. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J Am Chem Soc. 1991;113:4092–4096.
- Mahadevappa H, Martin SS, Kempaiah K, et al. Emerging roles of anacardic acid and its derivatives: a pharmacological overview. Basic Clin Pharmacol Toxicol. 2012;110:122–132.
- Rea AI, Schmidt JM, Setzer WN, et al. Cytotoxic activity of Ozoroa insignis from Zimbabwe. Fitoterapia. 2003;74:732–735.
- Kubo I, Kinst-Hori I, Yokokawa Y. Tyrosinase inhibitors from Anacardium occidentale fruits. J Nat Prod. 1994;57:545–551.
- Maorong S, Hasegawa I, Ishida Y, et al. Phenolic lipid ingredients from cashew nuts. J Nat Med. 2012;66:133–139.
- Kubo J, Jae RL, Kubo I. Anti-Helicobacter pylori agents from the cashew apple. J Agric Food Chem. 1999;47:533−537.
- Miriam C, Samir D, Erwin G, et al. 5-(12-Heptadecenyl)-resorcinol, the major component of the antifungal activity in the peel of mango fruit. Phytochemistry. 1986;25:1093−1095.