
ABSTRACT
(R)-2-amino-2-ethoxycarbonylsuccinimide (ASI-2) is a key intermediate used in the pharmaceutical industry and is valuable for the industrial synthesis of ranirestat, which is a potent aldose reductase inhibitor. ASI-2 was synthesized in a process combining chemical synthesis and bioconversion. Bioconversion in this study is a key reaction, since optically active carboxylic acid derivative ((R)-1-ethyl hydrogen 3-benzyloxycarbonylamino-3-ethoxycarbonylsuccinate, Z-MME-AE) is synthesized from a prochiral ester, diethyl 2-benzyloxycarbonylamino-2-ethoxycarbonylsuccinate, Z-MDE-AE, at a theoretical yield of 100%. Upon screening for microorganisms that asymmetrically hydrolyze Z-MDE-AE, Bacillus thuringiensis NBRC13866 was found. A novel esterase EstBT that produces Z-MME-AE was purified from Bacillus thuringiensis NBRC13866 and was stably produced in Escherichia coli JM109 cells. Using EstBT rather than porcine liver esterase (PLE), ASI-2 was synthesized with a 17% higher total yield by a novel method, suggesting that the esterase EstBT is a PLE substitute enzyme and therefore, may be of interest for future industrial applications.
Graphical abstract
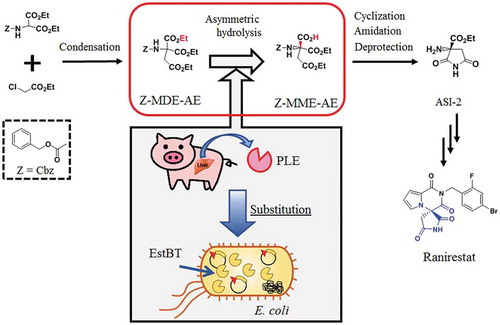
Production of a key intermediate ASI-2 of ranirestat using a recombinant enzyme overexpressed in Escherichia coli from the estBT gene, instead of the PLE enzyme.
ASI-2 [(R)-2-amino-2-ethoxycarbonylsuccinimide], which is a key intermediate used in the pharmaceutical industry, is valuable for the industrial synthesis of ranirestat ((R)-(−)-2-(4-bromo-2-fluorobenzyl)-1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazine-4-spiro-3′-pyrrolidine-1,2′,3,5′-tetrone) [Citation1]. Ranirestat is a potent aldose reductase inhibitor that was initially developed as a therapeutic agent for diabetic complications [Citation1]. Aldose reductase is the rate-limiting enzyme of the polyol pathway and catalyzes the reduction reaction from glucose to sorbitol. Increases in aldose reductase activity and accumulation of sorbitol in diabetic patients have been reported [Citation2]. Toyoda et al. [Citation3] have described that ranirestat reduced the area of stained glial fibrillary acidic protein and retinal thickness in Spontaneously Diabetic Torii (SDT) rats.
Several methods for synthesizing the key intermediate of ranirestat, ASI-2, using the optical resolution method have been reported [Citation1,Citation4–Citation7]. These methods have the drawback that recovery and racemization of enantiomer are needed after optical resolution. Kasai et al. [Citation8] have described a method of synthesizing ASI-2 without optical resolution. The method includes a hydrolysis step of a prochiral ester derivative to an optically active carboxylic acid derivative using a porcine liver esterase (PLE). There is an advantage that optically active carboxylic acid derivatives can be obtained with a theoretical yield of 100%; however, this method has the drawback of using PLE.
PLE, which is a type of the valuable esterases [Citation9–Citation13] used in manufacturing industries, is one of the most useful esterases for process chemistry and organic synthesis. Numerous reports [Citation6,Citation8,Citation14–Citation18] have highlighted various methods producing a wide range of optically active molecules using commercial PLE. However, commercial PLE has the drawback of being derived from porcine liver tissue extracts, which are obtained from an animal organ. In addition, commercial PLE has the drawback that its activity differs between various batches, since commercial PLE extracted from porcine liver tissue has different ratios of esterases [Citation19] and contains a plurality of isoenzymes (α, β, and γ-PLE) [Citation20].
To solve the problems associated with heterogeneous commercial PLE, many studies involving the production of recombinant γ-PLE have been reported. Brüsehaber et al. [Citation21] have described the expression of the γ-PLE gene with the Thioredoxin-tag (Trx-tag) in Escherichia coli. Böttcher et al. [Citation22] have described co-expression with the molecular chaperones GroEL and GroES in E. coli to produce γ-PLE. These methods have the drawback that the activity in whole cell is low for industrial applications. Böttcher et al. [Citation22] have also described that the γ-PLE gene was expressed with ompA (outer membrane protein A secretion signal) and relB (pectate lyase B signal sequence for periplasmic translocation) for periplasmic expression of the γ-PLE gene; however, the majority of γ-rPLE (recombinant γ-PLE) exists in an insoluble form as inclusion bodies in the cytoplasm. In the study with yeast as a host, Lange et al. [Citation23] have described that active γ-rPLE was secreted into the culture supernatant using recombinant Pichia pastoris expressing the γ-PLE gene. This method is limited by the fact that the activity of the culture supernatant is low for industrial applications.
Recently, in the manufacturing industries, environmental aspects are emphasized in addition to production costs. Many production processes using bioconversion reactions occur at ambient temperature and pressure [Citation24–Citation28], and are good for environmental conservation. In this study, we sought to identify an esterase, EstBT, that substitutes for PLE and constructed a scheme to synthesize the pharmaceutical intermediate ASI-2 in a process in which chemical synthesis and bioconversion are combined ().
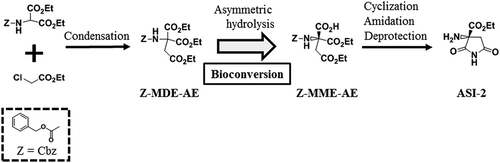
Figure 1. Scheme for the synthesis of ASI-2 via the bioconversion of Z-MDE-AE to Z-MME-AE.
Z-MDE-AE is synthesized by condensation. Z-MME-AE is synthesized by bioconversion of Z-MDE-AE. ASI-2 is synthesized from Z-MME-AE by cyclization, amidation, and deprotection. Z, Z-MDE-AE, Z-MME-AE, and ASI-2 indicate benzyloxycarbonyl group (Cbz), diethyl 2-benzyloxycarbonylamino-2-ethoxycarbonylsuccinate, (R)-1-ethyl hydrogen 3-benzyloxycarbonylamino-3-ethoxycarbonylsuccinate, and (R)-2-amino-2-ethoxycarbonylsuccinimide, respectively.
Materials and methods
Strains and plasmids
Bacillus thuringiensis NBRC3951, NBRC101235, and NBRC13866 were obtained from the National Institute of Technology and Evaluation (Tokyo, Japan). E. coli JM109 was obtained from Toyobo Co., Ltd. (Osaka, Japan). The E. coli expression vector pQE70 was obtained from Qiagen (Hilden, Germany).
Chemicals and enzymes
All chemicals were purchased from FUJIFILM Wako Pure Chemical Corp. (Osaka, Japan) and Sigma-Aldrich (St. Louis, MO, USA). PLE was purchased from Sigma-Aldrich. All restriction enzymes and DNA modification enzymes were purchased from Toyobo Co., Ltd. (Osaka, Japan) and New England Biolabs, Inc. (Ipswich, MA, USA). The synthetic oligonucleotides used in this study were purchased from Hokkaido System Science Co., Ltd. (Hokkaido, Japan).
Standard genetic manipulations and sequence analysis
Cloning was performed by standard genetic manipulation techniques [Citation29]. E. coli transformations were performed using the E. coli Transformation Buffer Set (Zymo Research Corp., Irvine, CA, USA). Genomic DNA was isolated using the Genomic DNA Buffer set and the Genomic-tip 500/G (Qiagen, Hilden, Germany). PCR fragments were prepared with KOD -plus- (Toyobo Co., Ltd.). DNA sequences of the plasmid inserts were determined using the primer-walking method described by Yamamura [Citation30]. Sequence assembly and analysis were performed using GENETYX Ver.12 (GENETYX Corp., Tokyo, Japan) and DNASIS Pro Ver.2.0 (Hitachi Software Corp., Tokyo, Japan).
Synthesis of diethyl 2-benzyloxycarbonylamino-2-ethoxycarbonylsuccinate (Z-MDE-AE)
A suspension of diethyl 2-benzyloxycarbonylaminomalonate (5.0 g), potassium carbonate (2.7 g), potassium iodide (0.27 g), and ethyl chloroacetate (2.6 g) in N,N-dimethylformamide (DMF, 50 mL) was stirred at 50°C for 1 h. The reaction mixture was cooled, and then poured into diluted hydrochloric acid, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and then dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure, and the residue was crystallized from ethyl acetate/n-hexane to obtain diethyl 2-benzyloxycarbonylamino-2-ethoxycarbonylsuccinate (Z-MDE-AE).
Screening of a bacterial library for asymmetric hydrolyzing activity of Z-MDE-AE
The actinomycetes and bacteria were inoculated into 5 mL of GPY broth (1% (w/v) glucose, 0.5% (w/v) Bacto peptone, and 0.3% (w/v) Bacto yeast extract) and incubated at 30°C with shaking for 3–7 days. The cells were harvested, freeze-dried, and suspended in 0.5 mL of 100 mM sodium phosphate buffer (pH 7.0) containing 0.1% (w/v) Z-MDE-AE and 10% (v/v) acetonitrile. The mixtures were incubated at 30°C with shaking for 72 h. The reactions were stopped by the addition of 1.5 mL of ethanol. Cell suspensions were centrifuged at 10,000 × g for 10 min at 4°C, and the resulting supernatants were analyzed by high-performance liquid chromatography (HPLC) as described below.
Analysis of reaction mixtures by HPLC
The hydrolysis rate of Z-MDE-AE and optical purity of the product 1-ethyl hydrogen 3-benzyloxycarbonylamino-3-ethoxycarbonylsuccinate were determined by HPLC on a CHIRALCEL OJ-RH (ϕ 4.6 × 150 mm; Daicel Corp., Tokyo, Japan). The mobile phase was acetonitrile/aqueous perchloric acid [pH 2.0, 30:70 (v/v)], the flow rate was 1.0 mL/min, the column temperature was 20°C, and the detection wavelength was at 254 nm.
Purification of an esterase EstBT
B. thuringiensis NBRC13866 cells were cultivated in GPY broth as described by Kozono et al. [Citation27]. An esterase EstBT was purified as described by Yamamura [Citation10]. B. thuringiensis NBRC 13866 was aerobically cultured in the GPY broth at 30°C for 16 h. Wet cells were collected from the culture broth by centrifugation (12,000 × g, 10 min) and suspended in 10 mM Tris buffer (pH 8.0). The suspension was disrupted by ultrasonication with an ultrasonic homogenizer US300 (Nihonseiki Kaisha Ltd., Tokyo, Japan) on ice, and then centrifuged at 14,000 × g for 20 min. The resulting supernatant was used as the crude extract. The crude extracts were added with ammonium sulfate at 20% (w/v) saturated concentration on ice. The mixture was stirred and centrifuged (12,000 × g, 10 min), and the supernatant was removed. The precipitates were suspended in 10 mM Tris buffer (pH 8.0) containing 1 mM dithiothreitol (DTT), and dialyzed against the same buffer. This solution was added with DEAE Sepharose Fast Flow (GE Healthcare UK Ltd., Buckinghamshire, England), the samples were mixed, and the mixture was filtered to collect an active solution. The solution was added with 3-[(3-chloroamidopropyl)dimethylammonio]-propanesulfonate (CHAPS) at a final concentration of 1% (w/v) and mixed. The mixture was subjected to treatment with an ultrasonic homogenizer for 30 min on ice, and then applied to DEAE Sepharose equilibrated with 10 mM Tris buffer (pH 8.0) containing 1 mM DTT and 1% (w/v) CHAPS (henceforth referred to as buffer A). The proteins were eluted with a linear gradient of 0 to 0.3 M NaCl in buffer A. The active fraction was dialyzed to remove CHAPS, combined with polyoxyethylene lauryl ether (Brij 35) at a final concentration of 0.05% (w/v) and NaCl at a final concentration of 0.3 M. Samples were then applied to Phenyl Sepharose High Performance (GE Healthcare UK Ltd., Buckinghamshire, England) equilibrated with 10 mM Tris buffer (pH 8.0) containing 0.05% (w/v) Brij 35, 1 mM DTT, and 0.3 M NaCl. The proteins were eluted with a linear gradient of 0 to 0.3 M NaCl in 10 mM Tris buffer (pH 8.0) containing 0.05% (w/v) Brij 35 and 1 mM DTT. The active fraction was dialyzed against buffer A, concentrated by ultrafiltration, and applied to Mono Q (GE Healthcare UK Ltd., Buckinghamshire, England) equilibrated with buffer A. The proteins were eluted with a linear gradient of 0 to 0.5 M NaCl in buffer A. The active fraction was applied to Benzamidine Sepharose 4 Fast Flow (GE Healthcare UK Ltd., Buckinghamshire, England) equilibrated with buffer A and then proteins were eluted with a linear gradient of 0 to 0.5 M NaCl. Protein purity was analyzed on SDS-PAGE (5–20% gradient gel) by the method described by Laemmli [Citation31] and was visualized by staining with Coomassie Brilliant Blue R250 and Silver Stain 2 kit purchased from FUJIFILM Wako Pure Chemical Corp. (Osaka, Japan). The SDS-PAGE standards (Broad) purchased from Bio-Rad Laboratories, Inc. (Hercules, CA, USA) were used as molecular weight markers. The purified enzyme was designated as EstBT. The amino acid sequence of the purified EstBT was determined by automated Edman degradation and the analysis was carried out at APRO Science Inc. (Tokushima, Japan).
Enzyme and protein assays
The reaction mixture consisting of 1% (w/v) Z-MDE-AE, 100 mM phosphate buffer (pH 7.0), 10% (v/v) acetonitrile, and enzyme was incubated at 30°C for 2 h. The reactions were stopped by addition of three volumes of ethanol and were centrifuged at 12,000 × g for 5 min at 4°C. The resulting supernatants were analyzed by high-performance liquid chromatography (HPLC) as described above. The activity for converting 1 µmol of Z-MDE-AE per 1 min was defined as 1 unit (U). Proteins were measured using a Bio-Rad Protein Assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with bovine serum albumin as a standard.
Construction of estBT expression vectors
The estBT gene that encodes EstBT, which contains a deletion of 40 amino acids at the N-terminus, was amplified using the following primers: P-F120: 5′-GTACAAAGCATGCAAAAGAAACCAGTCTCTTTAACGGAGCG-3′ (SphI site is underlined) and P-R1380: 5′-CTTTTGTTGTAGATCTTTTCTTTGTCGCATTGACCCA-3′ (BglII site is underlined), from the genomic DNA of B. thuringiensis NBRC13866 with a Mastercycler gradient thermal cycler (Eppendorf, Hamburg, Germany). The PCR fragment containing estBT was digested with SphI and BglII and then ligated into the SphI and BglII sites of pQE70 to generate the plasmid pBTEST38, which produces recombinant EstBT (rEstBT) with deletion of 40 amino acids at the N-terminus. To produce the full-length rEstBT (rEstBT-full), the full-length estBT was amplified using the following primers: P-F1: 5′-AAACAGCATGCAAAAAAAGAAATTACAAAAATCAGCAC-3′ (SphI site is underlined) and P-R1380: 5ʹ-CTTTTGTTGTAGATCTTTTCTTTGTCGCATTGACCCA-3ʹ (BglII site is underlined) from the genomic DNA of B. thuringiensis NBRC13866. The PCR fragment was digested with SphI and BglII, ligated into the SphI and BglII sites of pQE70, and designated as pBTEST12. The plasmids pBTEST38 and pBTEST12 produce rEstBT and rEstBT-full, respectively, with a His-tag attached to C-terminus.
Cultivation of recombinant E. coli expressed estBT and purification of rEstBT
Recombinant E. coli JM109 cells were inoculated into 5 mL of Luria-Bertani (LB) broth (1% Bacto tryptone, 0.5% Bacto yeast extract, and 1% NaCl) with ampicillin (100 μg/mL) and were incubated at 25°C with shaking for 16 h. Four milliliters of seed broth was transferred into 200 mL of LB broth with ampicillin (100 μg/mL) and isopropyl-β-D-thiogalactopyranoside (0.1 mM) and was incubated at 25°C with shaking for 24 h. With the Ni-NTA Fast Start Kit obtained from Qiagen (Hilden, Germany), the His-tagged rEstBT and the His-tagged rEstBT-full were purified from recombinant E. coli JM109 harboring the plasmids pBTEST38 and pBTEST12, respectively.
Activity measurements of recombinant E. coli JM109 cells
The reaction mixture consisting of 89 mM [3.5% (w/v) Z-MDE-AE, 100 mM buffer [standard buffer was phosphate buffer (pH 7.0)], and a portion of the cultured recombinant E. coli JM109 cells was incubated at 20–40°C (standard reaction condition was 25°C) for 2 h. The reaction was stopped by the addition of three volumes of ethanol and samples were centrifuged at 12,000 × g for 5 min at 4°C. The resulting supernatant was analyzed by HPLC as described above. The activity for converting 1 µmol of Z-MDE-AE per 1 min was defined as 1 U.
Producing (R)-1-ethyl hydrogen 3-benzyloxycarbonylamino-3-ethoxycarbonylsuccinate (Z-MME-AE) by bioconversion
The reaction mixture consisting of 89 mM [3.5% (w/v)] Z-MDE-AE, 100 mM phosphate buffer (pH 7.0), and 100 mU/mL enzyme (EstBT, rEstBT, PLE, or recombinant whole-cell) was incubated at 25°C. The pH of the reaction mixture was maintained at 7.0 with 28% NaOH automatically, since the pH decreases during the reaction. The reaction mixture was centrifuged at 12,000 × g for 5 min at 4°C to obtain the supernatant containing Z-MME-AE.
Synthesis of ASI-2 from Z-MME-AE
To cyclize Z-MME-AE, a solution of Z-MME-AE (1.8 g) in tetrahydrofuran (THF, 20 mL) was combined with triethylamine (0.96 mL) and isobutyl chloroformate (0.87 g) in this order at −15°C with stirring, and the mixture was stirred for 5 min. A solution of 25% aqueous ammonia (0.47 mL) was dropped into the reaction mixture at the same temperature. The reaction mixture was stirred at the same temperature for 1 h, and then poured into diluted hydrochloric acid. The mixture was extracted with ethyl acetate, and the extract was dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure, and the residue was subjected to silica gel column chromatography, in which elution was performed with n-hexane/ethyl acetate (1:1) for purification, and then crystallized from ethyl acetate/n-hexane. For amidation, a solution of the obtained crystals in dehydrated ethanol (75 mL) was added with sodium ethoxide (310 mg) on ice while stirring. The mixture was stirred at the same temperature for 2 h, then cold 1 mol/L hydrochloric acid was added, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and then dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure, and the residue was recrystallized from ethyl acetate/n-hexane. For deprotection, the obtained crystals were dissolved in ethanol (140 mL), the solution was added with 5% palladium/carbon (56 mg), and catalytic hydrogenation was performed at room temperature under a hydrogen atmosphere. After the catalyst was removed by filtration, the filtrate was concentrated under reduced pressure. The residue was recrystallized from ethanol to obtain ASI-2 as colorless crystals.
Nucleotide sequence accession number
The sequence of the estBT gene has been submitted to the DDBJ/EMBL/GenBank databases under accession number LC434454.
Results
Screening for Z-MDE-AE-asymmetric hydrolyzing microorganisms
E. coli is the most useful host for the expression of various genes and has many advantages, including easy handling, short generation times, various genetic tools, and high transformation efficiency. To express a gene in recombinant E. coli, prokaryotes were screened. A total of 495 strains from our bacterial library were used, consisting of 151 strains of actinomycetes and 344 strains of bacteria. Among the microbial strains, only one strain, B. thuringiensis NBRC13866, exhibited Z-MDE-AE-asymmetric hydrolyzing activity, and the optical purity of the product was 99.9% enantiomeric excess (ee) of the (R)-enantiomer.
Identification of a Z-MDE-AE-asymmetric hydrolyzing enzyme from B. thuringiensis NBRC13866
In enzyme purifications, the target enzyme can be efficiently purified by the addition of a surfactant to the buffer. Therefore, 12 different types of surfactants were screened. With two surfactants, CHAPS and Brij 35, the enzyme showed high specific activities in the active fraction in the purification steps. Without surfactant, no activity of EstBT was detected in fractions. For these reasons, CHAPS or Brij 35 was added to the buffer in the purification steps. On the other hand, in the enzyme reaction, the activity of EstBT was detected with no surfactant. For this reason, the activity measurements were performed without adding surfactants to the reaction solution.
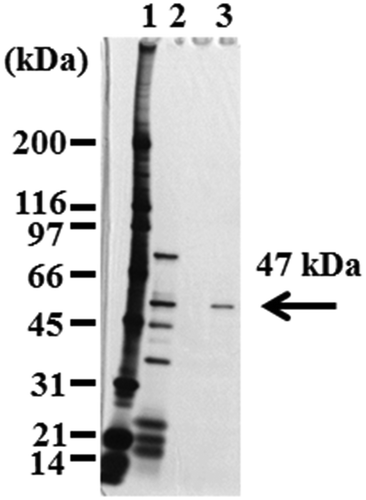
To identify the enzyme that hydrolyzes Z-MDE-AE to Z-MME-AE asymmetrically, the crude extracts of B. thuringiensis NBRC13866 were subjected to six purification steps (). In the first step, the ammonium sulfate precipitation step, the specific activity did not increase. However, when this step was omitted, EstBT could not be purified in next steps. For all purification steps, with the exception of the Phenyl Sepharose step, 1% (w/v) CHAPS was added to the buffer. In the Phenyl Sepharose step, 0.05% (w/v) Brij 35 was added to the buffer, since the specific activity of the active fraction increased. In this step, the addition of 1% (w/v) CHAPS hardly increased the specific activity. shows that proteins in the Benzamidine Sepharose step displayed a single protein band with a molecular weight of 49 kDa via SDS-PAGE. The purified protein was designated as EstBT. The specific activity of the purified EstBT was 160 mU/mg, and the optical purity of the product was 99.9% ee of the (R)enantiomer.
Table 1. Purification of esterase from Bacillus thuringiensis NBRC13866.
Figure 2. SDS-PAGE of EstBT from Bacillus thuringiensis NBRC13866.
Lane 1, size marker; lane 2, the active fraction of Mono Q; lane 3, the active fraction of Benzamidine Sepharose.
Amino acid and nucleotide sequence of EstBT
The amino acid sequence at the N-terminus of purified EstBT was determined by automated Edman degradation. Homology search for the obtained sequence (EKKPVSLTERTSLFF) was performed using the BLASTP 2.2.26 program (protein–protein Basic Local Alignment Search Tool, https://www.ddbj.nig.ac.jp/index-e.html) [Citation32]. The obtained sequence was homologous to α/β hydrolase (UniProt accession no. A0A243J741) derived from B. thuringiensis serovar pirenaica. Primers were prepared based on the sequence of the α/β hydrolase; the estBT gene that encodes EstBT was amplified by PCR from B. thuringiensis NBRC13866, and the sequence of the estBT gene was determined using the primer walking method [Citation30]. The estBT gene is 1380 bp in length and encodes a protein of 460 amino acid residues (). According to the TBLASTN 2.2.26 program (protein–translated nucleotide Basic Local Alignment Search Tool) [Citation32], EstBT is homologous to the α/β esterase (). EstBT contains a conserved serine protease motif (G-X-S-X-G) [Citation33], and the serine protease belongs to the superfamily of proteins with α/β hydrolase fold. Analysis of the amino acid sequence presumed from the sequence of the obtained estBT gene revealed that the N-terminal sequence (EKKPVSLTERTSLFF) of EstBT that was purified from B. thuringiensis NBRC13866 was not at the N-terminal presumed from the obtained estBT gene sequence but rather at 41 amino acid residues (). The predicted molecular weight from the amino acid sequence EKKPVSLTERTSLFF to the C-terminus of EstBT was 47 kDa, which is agreement with the results of SDSPAGE in .
Table 2. Features and identities between EstBT and known proteins.
Figure 3. Nucleotide sequence of estBT from Bacillus thuringiensis NBRC13866 and deduced amino acid sequence of full-length EstBT.
The conserved G-X-S-X-G motif of serine type proteases is boxed. The amino acid sequence at the N-terminus of EstBT purified from Bacillus thuringiensis NBRC13866 is underlined. Directing arrows indicate primers used to construct estBT expression vectors.

Expression of estBT in E. coli cells
For overexpression of the estBT gene in E. coli, two types of estBT expression vectors were constructed. The first is an expression vector that produces full-length EstBT with His-tag (rEstBT-full), designated as pBTEST12. The other is an expression vector that produces the His-tagged EstBT (rEstBT), which is truncated at 40 amino acid residues and resembles the EstBT purified from B. thuringiensis NBRC13866, designated as pBTEST38.
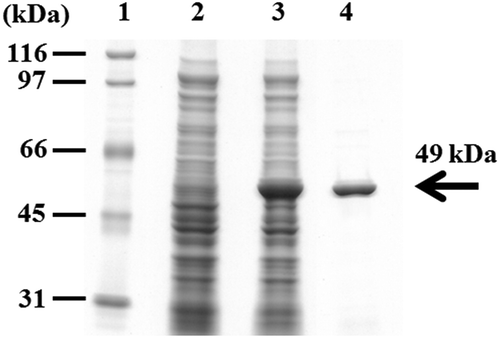
The plasmid pBTEST38 was transformed into E. coli JM109. rEstBT was overproduced in the recombinant E. coli JM109 and was detected in a soluble form with a molecular weight of 49 kDa on SDS-PAGE, which is in agreement with the predicted molecular weight (). The rEstBT was purified from the recombinant E. coli JM109 with the Ni-NTA Fast Start Kit (, lane 4). Analysis of the expression level of rEstBT by protein assay revealed 5.7 ± 0.5% rEstBT per dry cell weight (DCW). Moreover, the recombinant E. coli JM109 harboring the plasmid pBTEST12 also overproduced rEstBT-full. The rEstBT-full was detected in a soluble form with a molecular weight of 52 kDa on SDS-PAGE, which is in agreement with the predicted molecular weight. The production level of rEstBT-full was 5.5 ± 0.7% DCW.
Figure 4. SDS-PAGE of crude extracts from recombinant Escherichia coli JM109 expressing the estBT gene and the purified rEstBT.
Lane 1, size marker; lane 2, the crude extracts from recombinant E. coli JM109 harboring pQE-70; lane 3, the crude extracts from recombinant E. coli JM109 harboring pBTEST38; lane 4, the rEstBT purified from the recombinant E. coli JM109 harboring pBTEST38.
The relative activities of native EstBT, rEstBT, rEstBT-full, and PLE were determined to be 168 ± 8 mU/mg, 164 ± 6 mU/mg, 161 ± 12 mU/mg, and 288 ± 18 mU/mg, respectively (). The relative activities between native EstBT, rEstBT, and rEstBT-full were nearly identical, while that of PLE being approximately 1.7 times higher than those of the other enzymes. Additionally, the optical purities of (R)-enantiomer of native EstBT, rEstBT, rEstBT-full, and PLE were 99.9% ee, 99.9% ee, 99.9% ee, and 93.6% ee, respectively (). The optical purities between native EstBT, rEstBT, and rEstBT-full were the same, while those of EstBT, rEstBT, and rEstBT-full were higher than that of PLE.
Table 3. Comparison of the relative activity between EstBT, rEstBT, rEstBT-full, and PLE.
In the pH stability, incubation at pH 6, pH 7, and pH 8 for 2 h at 4°C did not reduce the enzyme activity of native EstBT, rEstBT, rEstBT-full, and PLE.
Characterization of recombinant E. coli JM109 producing rEstBT
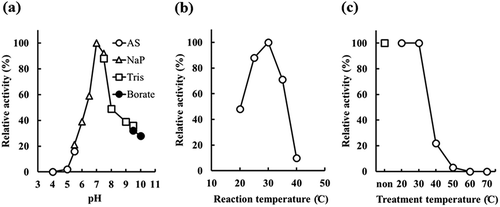
The optimal reaction conditions of recombinant E. coli JM109 producing rEstBT, which harbors the plasmid pBTEST38, were investigated to efficiently produce Z-MME-AE from Z-MDE-AE. The optimal pH of the activity of the recombinant E. coli JM109 was examined from pH 4.0 to 10.0 and was found to be 7.0 ()). The effects of reaction temperature and stable temperature were also examined. The maximum activity was observed at 30°C ()) and the stable temperature was 30°C or lower ()). Therefore, the bioconversion described below was performed at 25°C.
Figure 5. Determination of optimal pH and temperature by recombinant Escherichia coli JM109 producing rEstBT.
(a) Effect of pH. Activity was assayed under standard reaction conditions, except for the buffer. Open circle, open triangle, open square, and closed circle indicate acetic acid-sodium acetate buffer (AS), sodium phosphate buffer (NaP), Tris buffer (Tris), and borate buffer (borate), respectively. (b) Effect of reaction temperature. Activity was assayed under standard reaction conditions, except for temperature. (c) Temperature stability of rEstBT. Activity was assayed under standard reaction conditions. Reaction mixture was incubated for 30 min at 20–70°C before adding substrate. Open square indicates no temperature treatment. Open circle indicates temperature treatment at 20–70°C.
Bioconversion of Z-MDE-AE to Z-MMD-AE
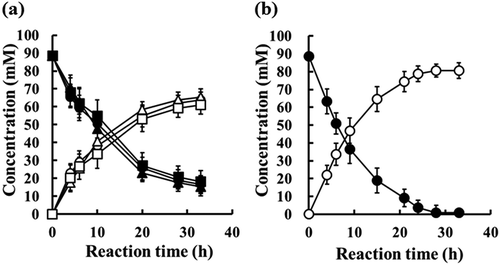
) shows bioconversion of Z-MDE-AE to Z-MMD-AE with 100 mU/mL of the enzymes EstBT, rEstBT, and PLE. The sequential changes in the production of Z-MME-AE with EstBT, rEstBT, and PLE were almost the same, and 89 mM [3.5% (w/v)] Z-MDE-AE was converted to approximately 61 mM Z-MME-AE (conversion rate 69%) in 33 h. On the other hand, ) shows bioconversion of Z-MDE-AE to Z-MMD-AE with 100 mU/mL recombinant whole-cell (wet-cell paste), which contains recombinant E. coli JM109 cells producing rEstBT. For 4 hours from the start of the reaction, the conversion rate with recombinant whole-cell was almost the same as those with enzymes of EstBT, rEstBT, and PLE. From the fourth hour onwards, the conversion rate with recombinant whole-cell was higher than those with EstBT, rEstBT, and PLE. In 33 h, 89 mM Z-MDE-AE was converted to approximately 81 mM Z-MME-AE (conversion rate 91%) with recombinant whole-cell.
Figure 6. Bioconversion of Z-MDE-AE to Z-MME-AE with enzymes at a final activity concentration of 100 mU/mL.
(a) Bioconversion with purified enzymes. Substrate is 89 mM Z-MDE-AE. Closed circle, closed triangle, and closed square indicate Z-MDE-AE concentrations of native EstBT, recombinant EstBT, and commercial PLE, respectively. Open circle, open triangle, and open square indicate Z-MME-AE concentrations of native EstBT, recombinant EstBT, and commercial PLE, respectively. Vertical bars indicate standard deviation (SD) from three independent experiments. (b) Bioconversion using recombinant Escherichia coli JM109 producing rEstBT. Substrate is 89 mM Z-MDE-AE. Closed circle and open circle indicate Z-MDE-AE and Z-MME-AE concentrations, respectively. Vertical bars indicate standard deviation (SD) from three independent experiments.
Synthesis of ASI-2 by chemical synthesis and bioconversion
ASI-2 was synthesized from diethyl 2-benzyloxycarbonylaminomalonate and ethyl chloroacetate (), which are readily and economically available starting materials. Via bioconversion with recombinant whole-cell producing rEstBT, ASI-2 was synthesized with a total yield of 55%. On the other hand, ASI-2 was synthesized with a total yield of 38% via bioconversion with PLE. Using recombinant whole-cell rather than PLE, ASI-2 was synthesized with a 17% higher total yield. Through the novel method using the PLE substitute enzyme, which is recombinant whole-cell producing rEstBT, ASI-2 was synthesized.
Discussion
PLE is one of several valuable esterases [Citation9–Citation13] used in manufacturing industries, and the most useful esterase for process chemistry, including the synthesis of ASI-2 [Citation8]. However, commercial PLE has the drawback that the activity is different between various batches, since commercial PLE extracted from porcine liver tissue has different ratios of esterases [Citation19] and contains a plurality of isoenzymes (α, β, and γ-PLE) [Citation20]. To address the problems associated with heterogeneous commercial PLE, many studies involving the production of recombinant γ-PLE have been reported. However, the activities of whole-cell are low for industrial applications.
We screened a PLE substitute enzyme that hydrolyzes Z-MDE-AE asymmetrically and is produced via recombinant techniques in E. coli. B. thuringiensis NBRC13866 hydrolyzed Z-MDE-AE asymmetrically and the optical purity of the product was 99.9% ee, while B. thuringiensis NBRC3951 and NBRC101235 from our bacterial library exhibited no hydrolysis activity of Z-MDE-AE. This might be due to differences in the membrane permeability of the substrate, expression levels of esterase gene, and substrate specificity of esterase among these strains. The freeze-dried cells and crude extracts of B. thuringiensis NBRC13866 hydrolyzed Z-MDE-AE, whereas wet cells of B. thuringiensis NBRC13866 exhibited no hydrolysis activity of Z-MDE-AE. Z-MDE-AE might have poor membrane permeability in B. thuringiensis NBRC 13866 since it is a gram-positive bacterial species.
To identify the enzyme that hydrolyzes Z-MDE-AE to Z-MME-AE asymmetrically, an enzyme purification protocol was performed. shows that proteins in the Benzamidine Sepharose step displayed the single protein band on the SDS-PAGE, suggesting that EstBT was purified from the crude extracts of B. thuringiensis NBRC13866 by six purification steps. In the first step of enzyme purification, which is the ammonium sulfate precipitation step, the specific activity did not increase, although this step was indispensable as biological substances other than proteins (e.g., lipids, sugars) might be removed at this time. The subsequent DEAE Sepharose step (through) was required. This is likely because many enzymes that interact strongly with DEAE were present in the active fraction of ammonium sulfate precipitation. In all purification steps, no activity of EstBT was detected without surfactant. Due to the absence of surfactant, it may be that the resin and EstBT directly interacted and the structure of EstBT was denatured [Citation10]. On the other hand, in the enzyme reaction, the activity of EstBT was detected with or without surfactant. This result suggests that EstBT is stable in aqueous solution with or without surfactant.
One of our research objectives is to obtain the gene of a PLE substitution enzyme that can be overexpressed in E. coli. To construct estBT expression vectors in E. coli, the nucleotide sequence of estBT was determined (). Homology search based on the determined sequence suggests that EstBT is a novel enzyme () and contains a conserved serine protease motif (G-X-S-X-G) [Citation33]. During enzyme purification (), EstBT was purified with Benzamidine Sepharose, which is a carrier for the purification of serine proteases, suggesting that EstBT is a serine protease.
Two kinds of the recombinant E. coli JM109 cells were constructed. The recombinant E. coli JM109 cells expressed the estBT gene in a soluble form, which is consistent with the predicted molecular weight. The production levels of rEstBT and rEstBT-full, which were produced in recombinant E. coli JM109 harboring pBTEST38 and pBTEST12, were 5.9 ± 0.5% DCW and 5.5 ± 0.8% DCW, respectively. These results suggest that the estBT gene was overexpressed in E. coli JM109.
The relative activities and the optical purities between native EstBT, rEstBT, rEstBT-full, and PLE were compared. The relative activities and the optical purities between native native EstBT, rEstBT, and rEstBT-full were nearly identical, suggesting that native EstBT, rPLE (truncated recombinant EstBT), and rEstBT-full (full-length recombinant EstBT) have the same activity. The relative activity of PLE was only 1.7 times higher than those of the other enzymes and the optical purity of PLE was lower than those of the other enzymes. These results suggest that rEstBT is the PLE substitute enzyme. However, we cannot explain the effect due to the difference of 40 amino acid residues between rEstBT and rEstBT-full. Due to the low solubility of Z-MDE-AE [<0.05% (w/v)], parameters (e.g., kcat, Km, or kcat/Km) of enzyme reaction could not be determined. In addition, substrate inhibition was not observed in 4% (w/v) Z-MDE-AE. This may be due to the low concentration of Z-MDE-AE dissolved in the reaction solution.
Bioconversion of the prochiral ester Z-MDE-AE to the optically active carboxylic acid Z-MME-AE is the key reaction in this process, since Z-MME-AE is synthesized at a theoretical yield of 100% (). For bioconversion of Z-MDE-AE to Z-MME-AE, the optimal reaction conditions of EstBT using recombinant E. coli JM109 producing rEstBT were investigated. The optimal pH, maximum reaction temperature, and stable temperature were 7.0, 30°C, and 30°C or less, respectively, which represent neutral pH and ambient temperature. These results suggest that EstBT is suitable for bioconversion.
Using enzymes (EstBT, rEstBT, PLE, and recombinant whole-cell producing rEstBT) at a final activity concentration of 100 mU/mL, bioconversion of Z-MDE-AE to Z-MME-AE was performed. For 4 hours from the start of the reaction, the Z-MME-AE concentrations among these enzymes in recombinant whole-cell were almost the same. These results suggest that there is almost no issue of membrane permeability of Z-MDE-AE and Z-MME-AE when recombinant whole-cell is used. From the fourth hour onwards, the Z-MME-AE concentration with recombinant whole-cell producing rEstBT was higher than that with the purified rEstBT. This result suggests that rEstBT is more stable in the cell than the purified rEstBT, and there is nearly no issue of compound stability of Z-MDE-AE and Z-MME-AE when the recombinant whole-cell is used. In addition, since cultured cells can be directly used for bioconversion, rather than purified enzyme, the synthesis process of Z-MME-AE is simplified. Moreover, the sequential changes in the production of Z-MME-AE with EstBT, rEstBT, and PLE were nearly identical. This result suggests that enzyme stabilities of EstBT and rEstBT are nearly identical to that of PLE. At 30 hours from the start of the reaction using enzymes, compounds presumed to be decomposition products of Z-MME-AE were detected, although the structures could not be determined. If the cause of this decomposition is clarified, it may be possible to synthesize Z-MME-AE via bioconversion at a conversion rate of 100%.
Numerous reports [Citation6,Citation8,Citation14–Citation18] have shown efficient methods to produce a wide range of optically active molecules using commercial PLE. In this study, we used recombinant whole-cell produced rEstBT, rather than PLE, to synthesize ASI-2 with a 17% higher total yield. This may be due to the fact that the yield was 20% higher in the bioconversion of Z-MDE-AE to Z-MME-AE (). In addition, Z-MME-AE may have been lost in the separation of Z-MME-AE from the reaction solution containing recombinant cells after bioconversion. Future studies to investigate the range of substrate specificity of rEstBT compared to PLE will be conducted using a wide range of molecules.
By making only effective optical isomers for pharmaceutical products, the effect is often enhanced and side effects are often reduced. As with PLE, EstBT is one of the useful enzymes in the pharmaceutical industry, since EstBT hydrolyzed Z-MDE-AE asymmetrically and the optical purity of the product was 99.9% ee.
In conclusion, a novel esterase EstBT, which is the PLE substitute enzyme, was identified and produced stably in E. coli. Using a novel method that combines chemical synthesis and bioconversion using recombinant whole-cell produced rEstBT, the pharmaceutical intermediate ASI-2 was synthesized efficiently.
Author contributions
Ei-Tora Yamamura, Kazuya Tsuzaki, and Shinji Kita conceived and designed the experiments. Shinji Kita investigated the patents on the ASI-2 manufacturing method. Ei-Tora Yamamura wrote the manuscript. Ei-Tora Yamamura and Kazuya Tsuzaki performed the experiments.
Acknowledgments
The authors would like to thank Dr. Masaji Kasai for the information on key bioconversion reactions. We would like to thank Dr. Tadashi Ogawa of Kyowa Pharma Chemical Co., Ltd., for the helpful information of compounds including substrates. We gratefully acknowledge Prof. Jun Ogawa of Kyoto University for his helpful advice regarding this manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Negoro T, Murata M, Ueda S, et al. Novel, highly potent aldose reductase inhibitors: (R)-(−)-2-(4-bromo-2-fluorobenzyl)-1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazine-4-spiro-3ʹ-pyrrolidine-1,2ʹ,3,5ʹ-tetrone (AS-3201) and its congeners. J Med Chem. 1998;41:4118–4129.
- Hamada Y, Kitoh R, Raskin P. Crucial role of aldose reductase activity and plasma glucose level in sorbitol accumulation in erythrocytes from diabetic patients. Diabetes. 1991;40:1233–1240.
- Toyoda F, Tanaka Y, Ota A, et al. Effect of ranirestat, a new aldose reductase inhibitor, on diabetic retinopathy in SDT rats. J Diabetes Res. 2014; article ID 672590. 1–7. doi:10.1155/2014/672590
- Tanaka D, inventor; Dainippon Sumitomo Pharma Co., Ltd., assignee. 3-Hydrazino-2,5-dioxopyrrolidine-3-carboxylates, process for production of the same, and use of the same. United States patent US 8,003,808 B2. 2011 Aug 23.
- Tanaka D, Negoro T, inventors; Dainippon Sumitomo Pharma Co., Ltd., assignee. Optically active 3-amino-2,5-dioxopyrrolidine-3-carboxylate, process for production of the compound, and use of the compound. United States patent US 8,058,456 B2. 2011 Nov 15.
- Kudo Y, Yamada O, inventors; Nissan Chemical Industries, Ltd., Dainippon Sumitomo Pharma Co., Ltd., assignees. Method for producing optically active succinimide compound. United States patent US 7,994,342 B2. 2011 Aug 9.
- Inagaki T, Yamakawa Y, inventors; Dainippon Sumitomo Pharma Co., Ltd., and Katayama Seiyakusyo Co., Ltd., assignees. Succinic acid diester derivative, process for production thereof, and use of the derivative in the production of pharmaceutical preparation. United States patent US 8,030,486 B2. 2011 Oct 4.
- Kasai M, Kita S, Ogawa T, et al. inventors; Dainippon Sumitomo Pharma Co., Ltd., and Kyowa Hakko Bio Co., Ltd., assignees. Process for producing optically active succinimide derivatives and intermediates thereof. United States patent US 8,633,001 B2. 2014 Jan 21.
- Hayashi Y, Onaka H, Itoh N, et al. Cloning of the gene cluster responsible for biosynthesis of KS-505a (longestin), a unique tetraterpenoid. Biosci Biotechnol Biochem. 2007;71:3072–3081.
- Yamamura ET, Kita S. A novel method of producing the pharmaceutical intermediate (R)-2-chloromandelic acid by bioconversion. Biosci Biotechnol Biochem. 2019;83:309–317.
- Okazaki R, Kiyota H, Oritani T. Enzymatic resolution of (±)-epoxy-β-cyclogeraniol, a synthetic precursor for abscisic acid analogs. Biosci Biotechnol Biochem. 2000;64:1444–1447.
- Kanjanavas P, Khuchareontaworn S, Khawsak P, et al. Purification and characterization of organic solvent and detergent tolerant lipase from thermotolerant Bacillus sp. RN2. Int J Mol Sci. 2010;11:3783–3792.
- Park EY, Sato M, Kojima S. Lipase-catalyzed biodiesel production from waste activated bleaching earth as raw material in a pilot plant. Bioresour Technol. 2008;99:3130–3135.
- Bornscheuer UT, Kazlauskas RJ. Hydrolases in organic synthesis -regio- and stereoselective biotransformations. 2nd ed. Weinheim: Wiley-VCH; 2006.
- Faber K. Biotransformations in organic chemistry. 5th ed. Berlin, Heidelberg, New York: Springer; 2004.
- de Maria PD, Kossmann B, Potgrave N, et al. Improved process for the enantioselective hydrolysis of prochiral diethyl malonates catalyzed by pig liver esterase. Synlett. 2005;11:1746–1748.
- Lam LKP, Hui RAHF, Jones JB. Enzymes in organic synthesis. 35. Stereoselective pig liver esterase catalyzed hydrolyses of 3-substituted glutarate diesters. Optimization of enantiomeric excess via reaction conditions control. J Org Chem. 1986;51:2047–2050.
- Bornscheuer U, Hummel A, Böttcher D, et al. inventors; Enzymicals AG, assignee. Isoforms of pig liveresterase. United States patent US 8,304.223 B2. 2012 Nov 6.
- Seebach D, Eberle M. Enantioselective cleavage of meso-nitrodiol diacetates by an esterase concentrate from fresh pig liver: preparation of useful nitroaliphatic building blocks for EPC syntheses. Chimia (Aarau). 1986;40:315–318.
- Faber D, Jencks WP. Different forms of pig liver esterase. Arch Biochem Biophys. 1980;203:214–226.
- Brüsehaber E, Schwiebs A, Schmidt M, et al. Production of pig liver esterase in batch fermentation of E. coli Origami. Appl Microbiol Biotechnol. 2010;86:1337–1344.
- Böttcher D, Brüsehaber E, Doderer K, et al. Functional expression of the γ-isoenzyme of pig liver carboxyl esterase in Escherichia coli. Appl Microbiol Biotechnol. 2007;73:1282–1289.
- Lange S, Musidlowska A, Schmidt-Dannert C, et al. Cloning, functional expression, and characterization of recombinant pig liver esterase. Chembiochem. 2001;2:576–582.
- Menon V, Rao M. Trends in bioconversion of lignocellulose: biofuels, platform chemicals & biorefinery concept. Prog Energy Combust Sci. 2012;38:522–550.
- Takeuchi M, Kishino S, Park SB, et al. Efficient enzymatic production of hydroxy fatty acids by linoleic acid Δ9 hydratase from Lactobacillus plantarum AKU 1009a. J Appl Microbiol. 2016;120:1282–1288.
- Yamamura ET. Bioconversion of pyridoxine to pyridoxamine through pyridoxal using a Rhodococcus expression system. J Biosci Bioeng. 2019;127:79–84.
- Kozono I, Mihara K, Minagawa K, et al. Engineering of the cytochrome P450 monooxygenase system for benzyl maltol hydroxylation. Appl Microbiol Biotechnol. 2017;101:6651–6658.
- Kato Y, Tsuda T, Asano Y. Nitrile hydratase involved in aldoxime metabolism from Rhodococcus sp. strain YH3-3 purification and characterization. Eur J Biochem. 1999;263:662–670.
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. New York (NY): Cold Spring Laboratory; 1989.
- Yamamura ET. Construction of Rhodococcus expression vectors and expression of the aminoalcohol dehydrogenase gene in Rhodococcus erythropolis. Biosci Biotechnol Biochem. 2018;82:1396–1403.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685.
- Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402.
- Obst M, Oppermann-Sanio FB, Luftmann H, et al. Isolation of cyanophycin-degrading bacteria, cloning and characterization of an extracellular cyanophycinase gene (cphE) from Pseudomonas anguilliseptica strain BI. The cphE gene from P. anguilliseptica BI encodes a cyanophycinhydrolyzing enzyme. J Biol Chem. 2002;277:25096–25105.
- Lapidus A, Goltsman E, Auger S, et al. Extending the Bacillus cereus group genomics to putative food-borne pathogens of different toxicity. Chem Biol Interact. 2008;171:236–249.
- Johnson SL, Daligault HE, Davenport KW, et al. Complete genome sequences for 35 biothreat assay-relevant bacillus species. Genome Announc. 2015;3:e00151–15.