
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Pulmonary hypertension (PH) is a life-threatening lung disease. PH with concomitant lung diseases, e.g., idiopathic pulmonary fibrosis, is associated with poor prognosis. Development of novel therapeutic vasodilators for treatment of these patients is a key imperative. We evaluated the efficacy of dual activation of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) using an active, small-molecule phosphodiesterase (PDE4)/PDE5 dual inhibitor (Compound A). Compound A increased both cAMP and cGMP levels in WI-38 lung fibroblasts and suppressed the expressions of type-1 collagen α1 chain and fibronectin. Additionally, compound A reduced right ventricular weight/left ventricular weight+septal weight ratio, brain natriuretic peptide expression levels in right ventricle, C─C motif chemokine ligand 2 expression levels in lung, and plasma surfactant protein D. Our data indicate that dual activation of cAMP/cGMP pathways may be a novel treatment strategy for PH.
Graphical abstract
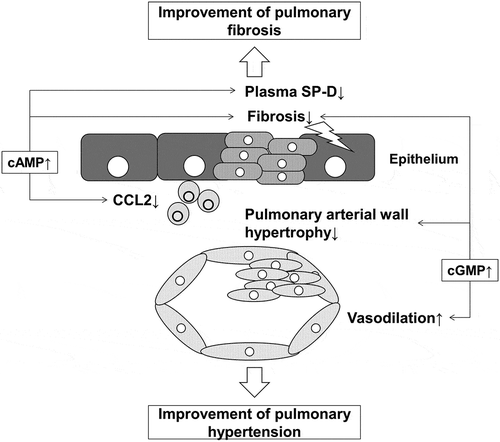
Elevated cAMP suppresses profibrotic and inflammatory responses. Elevated cGMP enhances vasodilation and suppresses proliferation of smooth muscle cells.
Introduction
Pulmonary hypertension (PH) is a life-threatening lung disease that affects an estimated 100,000–200,000 individuals worldwide [Citation1]. Despite the availability of several therapeutics, such as endothelin receptor antagonists (ERAs), phosphodiesterase (PDE) 5 inhibitors, prostaglandin (PG) I2 analogs, and soluble guanylate cyclase (sGC) activators [Citation1], life expectancy after a diagnosis of PH is in the vicinity of 6–7 years [Citation2].
PH is divided into five categories based on the etiology: Group 1, PH due to pulmonary arterial remodeling; Group 2, PH due to left heart disease; Group 3, PH due to lung diseases, such as chronic obstructive pulmonary disease or idiopathic pulmonary fibrosis (IPF); Group 4, PH due to chorionic thromboembolic pulmonary hypertension; and Group 5, PH with unclear multifactorial hypertension [Citation3]. ERA, PGI2 analogs, and PDE5 inhibitors are approved for treatment of Group 1 PH [Citation3], while sGC activators are approved for patients with Group 1 and Group 4 PH [Citation4]. Despite recent advances in drug development for PH, treatment options available for Group 3 PH remain limited, and PH with IPF continues to be associated with poor prognosis. IPF is a life-threatening condition that manifests as chronic cough, exertional dyspnea, and acute respiratory failure [Citation5]. Approximately 10% of all patients with IPF present with PH at initial IPF diagnosis and 32%–50% of all patients with IPF eventually develop PH. Unfortunately, vasodilators have limited therapeutic efficacy in patients with PH-IPF. A previous clinical trial of ERA in IPF patients with and without PH was prematurely terminated due to disease progression in the treatment group [Citation6].
In contrast, PDE5 inhibitors were shown to improve survival rates of PH patients with respiratory diseases [Citation7]. PDE5 inhibitors induce production of cyclic guanosine monophosphate (cGMP), which in turn induces vasodilation in the pulmonary vasculature. Moreover, PDE5 inhibitors were shown to improve respiratory function scores (an index of severity of PH) in IPF patients with right ventricular hypertrophy [Citation8]. Thus, PDE5 inhibitors improve ventilation/perfusion matching in PH-IPF by reducing pulmonary artery pressure [Citation9].
PH-IPF is the result of a vicious cycle of PH, epithelial cell injury, fibroblast activation, and inflammatory cell response [Citation10]. Thus, an ideal therapeutic agent for PH-IPF should also attenuate tissue injury. Previous research has shown that activation of cyclic adenosine monophosphate (cAMP) facilitates tissue protection via anti-inflammatory, epithelioprotective, and anti-fibrotic effects [Citation11,Citation12]. Therefore, dual activation of the cAMP and cGMP pathways is a potential approach for treatment of PH-IPF. In this study, we aimed to evaluate the effects of dual activation of the cAMP/cGMP pathway on inflammatory response, epithelial protection, and pulmonary arterial hypertension using a rat model of monocrotaline (MCT)-induced PH (MCT rat).
Materials and methods
In vitro cAMP and cGMP assay
For cAMP and cGMP assay, WI-38 human lung fibroblasts were suspended in Eagle’s minimal essential medium (E-MEM) (Thermo Fisher Scientific, MA, USA) containing 10% heat-inactivated fetal bovine serum (FBS) and seeded at 50,000 cells/500 μL in each well of a 24-well plate or at 250,000 cells/2.5 mL in each well of a 6-well plate. To activate cAMP/cGMP pathway, we used a small molecule compound that has an inhibitory effect on both PDE4 and PDE5 (Compound A, synthesized by Takeda Pharmaceutical Company Limited). Twenty-four hours after seeding, cells were treated with compound A (0.001–10 μM) dissolved in Hanks’ balanced salt solution (Thermo Fisher Scientific) containing 0.1% bovine serum albumin (BSA) and 5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (n = 3). After 30 min, cells were stimulated with 1 μM forskolin for 30 min for cAMP measurement or 2 h for cGMP measurement. Cells were lysed by lysis buffer provided by the manufacturer. Intracellular cAMP and cGMP levels were measured using the cAMP XP assay kit (Cell Signaling Technology, MA, USA) or the cGMP enzyme-linked immunosorbent assay (ELISA) kit (Enzo Biochem, Farmingdale, NY).
Compound A is a chemically synthesized, single, small molecule compound. The chemical structure of compound A was identified, which can be disclosed in future. The reproduce study using compound A can be conducted in future. The 50% inhibition concentration (IC50) for human PDE4B, 4D and PDE5A proteins measured using scintillation proximity assay (SPA) (GE Healthcare, IL, USA) [Citation13] are shown in . Either recombinant PDE4B, PDE4D or PDE5A (BPS biosciences Inc, CA, USA) was pre-treated with compound A dissolved in assay buffer (50 mM HEPES-NaOH, 8.3 mM MgCl2, 1.7 mM EGTA, 0.1% BSA, pH 7.4) for 30 min. Then, either [3H] cAMP or [3H] cGMP was added to the mixture and the PDE reaction was conducted for 60 min. After reaction for 60 min, yttrium SPA beads containing zinc sulfate was added to terminate the PDE reaction. The radioactivity of samples was measured using scintillation counter (PerkinElmer Inc, MA, USA). IC50 values were obtained by fitting the results to the following four-parameter logistic equation:
Table 1. Inhibition of PDE4 and PDE5 by compound A.
where A is the minimum y value, B is the maximum y value, C is Log(IC50) value, and D is the slope factor.
In vitro gene expression assay
For gene expression assay, WI-38 human lung fibroblasts were suspended in E-MEM containing 10% heat-inactivated FBS and seeded on 24-well plates at 50,000 cells/500 μL/well. Twenty-four hours after seeding, the medium was replaced with E-MEM containing 0.5% FBS. After 24 h, cells were treated with compound A (0.001–10 μM) (n = 3). After 1 h, cells were treated with transforming growth factor beta (TGF-β; 3 ng/mL) and forskolin (1 μM). After 24 h incubation, total RNA was extracted using the RNeasy kit (Qiagen, Hilden, Germany). cDNA was synthesized using the high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific), and target gene mRNA levels were measured using TaqMan gene expression assays (Thermo Fisher Scientific) and a TaqMan PCR device (7900HT, Thermo Fisher Scientific). Gene expression levels were normalized to the mRNA expression level of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). For primer and probe sets, Assay IDs for GAPDH, type-1 collagen α1 chain (COL1A1), fibronectin (FN1), PDE3A, PDE3B, PDE4A, PDE4B, PDE4C, PDE4D, PDE5A, PDE7A, PDE7B, and PDE10A were Hs02758991g1, Hs00164004m1, Hs00365052m1, Hs01012698m1, Hs00265322m1, Hs00183479m1, Hs00963643m1, Hs00971865m1, Hs01579625m1, Hs00153649m1, Hs00300285m1, Hs01054008m1, and Hs01098924m1, respectively.
Pharmacokinetics (PK)/pharmacodynamics (PD) analysis
The protocol for animal experiment was approved by the Institutional Animal Care and Use Committee of Takeda Pharmaceutical Company Limited. Seven-week-old Wistar rats (CLEA Japan, Tokyo, Japan) were anesthetized with isoflurane (3%) and administered 60 mg/kg MCT (WAKO Pure Chemical Industries, Tokyo, Japan) dissolved in saline via subcutaneous injection. One week after MCT administration, compound A (10 mg/kg) was orally administered to MCT rats who were sacrificed at 0.5, 1, 2, 4, 8, and 24 h after dosing (n = 4). Blood samples were collected and centrifuged to obtain plasma specimens. Plasma concentrations of compound A were measured using liquid chromatography/tandem mass spectrometry. Skeletal muscle and lung tissues were collected to measure cAMP and cGMP levels, respectively. Briefly, tissues were homogenized and deproteinized in 6% trichloroacetic acid, followed by centrifugation and washing with diethyl ether. The water layer was freeze-dried and dissolved in lysis buffer provided in the cAMP XP assay and cGMP ELISA kits.
In vivo pharmacology studies in the MCT rat
Seven-week-old Wistar rats were anesthetized with isoflurane (3%) and administered MCT (60 mg/kg) (WAKO Pure Chemical Industries) dissolved in saline via subcutaneous injection. One week after administration of MCT, blood samples were collected from the tail vein and plasma level of surfactant protein D (SP-D) was measured using the SP-D ELISA kit (YAMASA CORPORATION, Chiba, Japan) to monitor the extent of lung injury. Briefly, plasma samples were diluted in the buffer provided in the kit, and SP-D levels were measured according to the manufacturer’s instructions. Sildenafil (20 or 60 mg/kg q.d.) and compound A (1, 3, or 10 mg/kg, q.d.) were orally administered for two weeks (n = 4–12/treatment). At the end of the treatment, rats were sacrificed 2 or 6 h after the final administration. Blood samples were collected and centrifuged to obtain plasma specimens for measurement of SP-D. The ratio of right ventricular weight to that of left ventricle plus septum (RV/LV+S) was measured as an index of pulmonary arterial pressure. Left ventricle and lung were collected and stored in the RNAlater solution (Qiagen) for gene expression analyses. Total RNA was extracted from tissue using the RNeasy kit. High-Capacity cDNA reverse transcription kit was used for cDNA synthesis. Target gene mRNA levels were measured using TaqMan gene expression assays and the TaqMan 7900HT PCR device. The target gene expression levels were normalized to the mRNA expression level of GADPH. Assay IDs for GADPH, C─C motif chemokine ligand 2 (Ccl2), brain natriuretic peptide (BNP) were Rn01775763_g1, Rn00580555_m1, and Rn00580641_m1, respectively. For histopathological examination, lung samples were fixed in 10% paraformaldehyde and sectioned and stained with hematoxylin-eosin and Elastica van Gieson stains. For measurement of PK parameters after repeated administration, blood samples were collected from the tail vein at 0.5, 1, 2, 4, 8, and 24 h after final administration.
Statistical analysis
Student’s t-test was used to evaluate differences between the two groups. Williams’ test was used to evaluate dose-dependent effects and Pearson’s correlation coefficient was used to measure the strength of the association between two groups. P values less than 0.05 were considered as statistically significant.
Results
Dual activation of cAMP/cGMP pathway in vitro
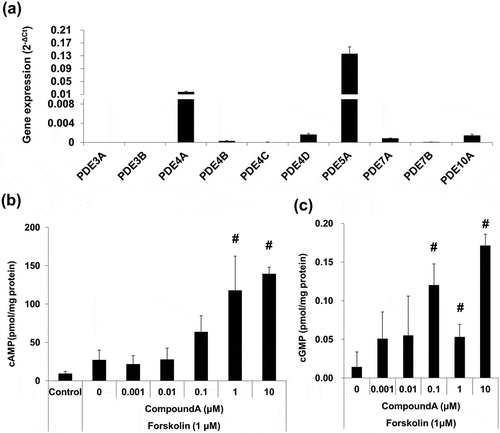
The effect of compound A on cAMP and cGMP production was evaluated using the WI-38 cell line. Among the PDE genes, WI-38 cells expressed PDE4A and PDE5A, whereas the expression levels of PDE4D, PDE7A, and PDE10A were lower. Expression levels of PDE3A, PDE3B, PDE4B, PDE4C, and PDE7B were near or below the limit of detection ()). In the presence of forskolin at concentrations of 1 μM, compound A significantly elevated cAMP levels in WI-38 cells ()). At 10 μM, compound A led to a 510.6% increase in the cAMP level as compared to that observed after the treatment with 1 μM forskolin alone. In addition, compound A also significantly elevated cGMP levels in WI-38 fibroblasts ()). Significant increase in cGMP levels was observed in cells treated with compound A at concentrations ≥0.1 μM. For example, compound A at 10 μM led to a 1200.7% increase in cGMP in 1 μM forskolin-co-treated cells as compared to that after the treatment with 1 μM forskolin alone. Overall, these findings demonstrated that compound A induced cAMP/cGMP pathway in lung fibroblasts.
Figure 1. Elevation of cAMP and cGMP by compound A in the presence of forskolin in WI-38 human lung fibroblast cell line.
(a) Expression of PDEs in WI-38 lung fibroblasts. Gene expression levels were normalized to that of GAPDH. (b, c) Measurement of intracellular cAMP and cGMP levels; (b) Treatment with compound A for 30 min led to an increase in cAMP in WI-38 cells stimulated with forskolin. (c) Treatment with compound A for 2 h led to an increase in cGMP in WI-38 cells treated with compound A for 2 h. C: control, mean ± SD, n = 3, #p ≤ 0.05 vs. control by two-tailed Williams’ test.
Suppression of TGF-β-induced profibrotic gene expression by dual activation of cAMP/cGMP pathway
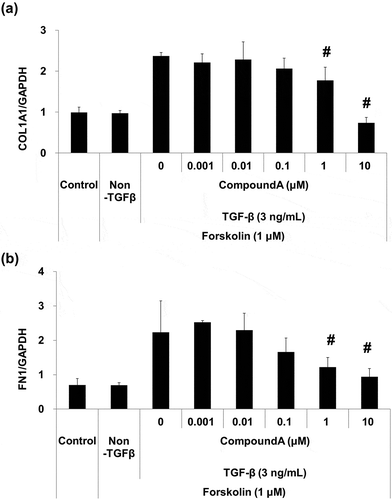
To evaluate the anti-fibrotic effect of the dual activation of cAMP/cGMP pathway, we assessed the profibrotic gene expression levels in WI-38 cells. As shown in , TGF-β led to increased expression levels of COL1A1 and FN1, while no significant change was observed in cells co-treated with 1 μM forskolin. However, in the presence of forskolin, compound A suppressed the TGF-β-induced COL1A1 and FN1 gene expression in a concentration-dependent manner (). Compound A significantly reduced COL1A1 and FN1 gene expression at concentrations ≥1 μM.
Figure 2. Inhibitory effect of compound A on TGF-β-induced profibrotic gene expression in WI-38 human lung fibroblast cell line.
(a) COL1A1 expression; (b) FN1 expression; WI-38 cells were treated with compound A for 1 h and stimulated by forskolin/TGF-β in the presence of compound A for 24 h. Mean ± SD, n = 3, #p ≤ 0.05 vs. forskolin/TGF-β by two-tailed Williams’ test. COL1A1, type-1 collagen α1 chain; FN1, fibronectin; TGF-β, transforming growth factor-β.
In vivo activation of the cAMP/cGMP pathway by compound A
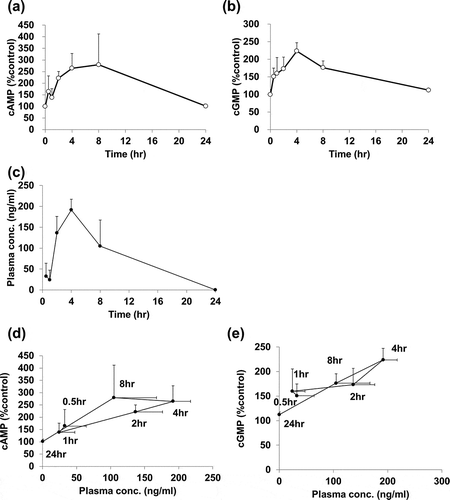
To confirm dual activation of cAMP/cGMP pathway by compound A in vivo, we measured the levels of cAMP and cGMP in MCT rat. Changes in cAMP levels were evaluated in skeletal muscle based on a previous study, which showed that systemic administration of PDE4 and PDE5 inhibitors increases cAMP levels in skeletal muscle and increases cGMP levels in lung, respectively [Citation14]. Consistent with those findings, compound A at a dose of 10 mg/kg increased cAMP in skeletal muscle and cGMP in lung (). Correlation between plasma concentrations of compound A, cAMP, and cGMP level was also assessed by evaluating the PK profile of compound A in MCT rats. Compound A at 10 mg/kg resulted in a peak plasma concentration of 191.7 ng/mL at 4 h, which was sustained for at least 8 h, and was not detected in plasma 24 h after administration ()). The cAMP and cGMP levels showed a correlation with plasma concentration of compound A ().
Figure 3. In vivo activation of cAMP/cGMP pathways by compound A in a rat model of monocrotaline-induced pulmonary hypertension.
Compound A (10 mg/kg) was orally administered to MCT rats. Plasma, lung, and skeletal muscle samples were collected at 0.5–24 h following administration. (a) Elevation of cAMP in skeletal muscle; (b) Elevation of cGMP in lung; (c) Plasma concentration-time profiles of compound A after oral administration; (d) Relationship between plasma concentration of compound A and cAMP level. (e) Relationship between plasma concentration of compound A and cGMP level. Mean ± SD, n = 4. MCT, monocrotaline.
Effects of activation of cGMP pathway and dual activation of cAMP/cGMP pathway on right ventricular hypertrophy, pro-inflammatory response, and epithelial injury in MCT rat
To evaluate the effect of cGMP pathway and cAMP/cGMP pathway in MCT rat, Sildenafil and compound A were administered to MCT rat for two weeks, starting one week after administration of MCT. Indices of pulmonary arterial pressure, RV/LV+S, and right ventricular BNP gene expression were elevated in the MCT rats (). Sildenafil (60 mg/kg) significantly suppressed the increase in RV/LV+S and BNP expression (). In addition, compound A significantly suppressed RV/LV+S and BNP expression at doses of 3 and 10 mg/kg, respectively (). Moreover, lung Ccl2 expression, an index of inflammatory response which was not affected by sildenafil treatment, was decreased by compound A ()). SP-D, an index of epithelial injury, was elevated in MCT-treated rats in control as well as sildenafil- and compound A (1 and 3 mg/kg) treatment groups. In contrast, no increase in SP-D was observed in the rats treated with compound A (10 mg/kg) ()). Skeletal muscle cAMP content was significantly increased 6 h after the final administration of compound A (10 mg/kg) ()). In addition, lung cGMP content was increased in a dose-dependent manner by sildenafil as well as compound A ()). Furthermore, a dose-dependent increase in plasma compound A concentration was observed after two weeks of repeated administration ()).
Figure 4. Effect of compound A on the index of pulmonary arterial pressure and epithelium protection in a rat model of monocrotaline-induced pulmonary hypertension.
Compound A and sildenafil were orally administered to MCT rats for two weeks. (a) Effect on RV/LV+S ratio. (b) Effect on BNP gene expression in the right ventricle. (c) Effect on Ccl2 gene expression in the lung. (d) Effect on plasma concentration of surfactant protein D (SP-D). Mean ± SD. n = 4–12. #p ≤ 0.05 vs. vehicle group by two-tailed Williams’ test. *p ≤ 0.05 vs. Pre value by Student’s t-test. RV/LV+S, ratio of right ventricular weight to that of left ventricle plus septum; BNP, brain natriuretic peptide; Ccl2, C─C chemokine 2; SP-D, surfactant protein D.
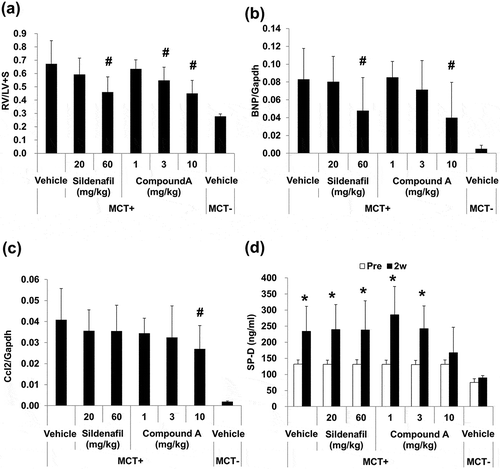
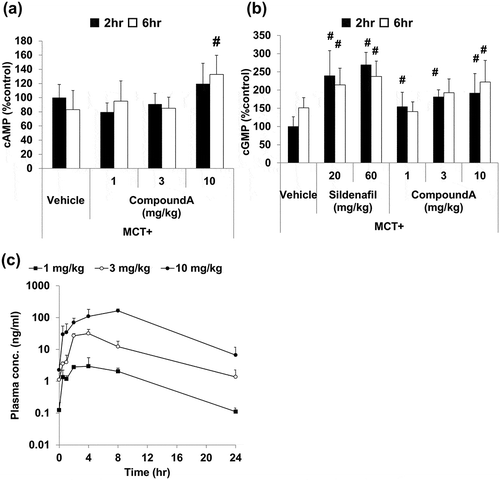
Figure 5. Levels of cAMP and cGMP and pharmacokinetic parameters after repeated dosing of compound A.
Compound A (1, 3, or 10 mg/kg, q.d.) and sildenafil (20 or 60 mg/kg, q.d.) were orally administered to MCT rats for two weeks. Skeletal muscle and lung samples were collected 2 and 6 h after final administration. Plasma samples were collected at 0.5, 1, 2, 4, 8, and 24 h after the final administration. (a) Elevation of cAMP in the skeletal muscle. (b) Elevation of cGMP in the lung. (c) Plasma concentration-time profile of compound A. Mean ± SD. n = 4–12. #p ≤ 0.05 vs. vehicle group by two-tailed Williams’ test.
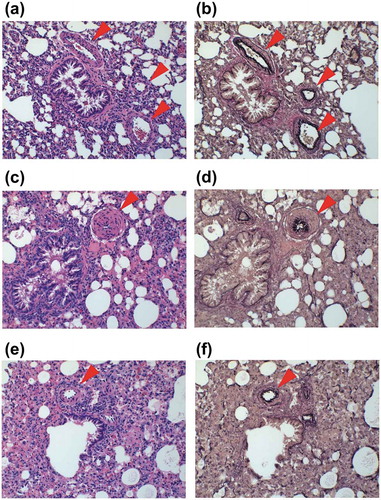
Histopathological analysis was conducted to evaluate pulmonary arterial hypertrophy using one representative sample from each of the MCT, compound A, and control groups. Mild to modest pulmonary arterial hypertrophy due to the proliferation of smooth muscle cells, which was absent in the control group (), was observed in the MCT group (). In contrast, the level of pulmonary artery hypertrophy was milder in the compound A group ().
Figure 6. Representative micrographs of the pulmonary arterial wall.
(a, b) Control group. (c, d) Groups treated with monocrotaline (MCT) (60 mg/kg) or vehicle. Samples were collected three weeks after MCT administration. (e, f) Group treated with MCT (60 mg/kg) and compound A (10 mg/kg). Compound A was administered for two week from one week after MCT administration. Samples were stained by hematoxylin and eosin (a, c, e) and Elastica van Gieson stain (b, d, f). Arrowheads indicate pulmonary arterial wall.
Discussion
IPF is a life-threatening disorder and concurrent presence of PH worsens the prognosis of IPF. Despite recent advances in the development of drugs for PH and IPF, efficient therapeutic approaches have not been established. In previous studies, PDE5 inhibitors were shown to ameliorate disease score and prognosis of patients with PH-IPF [Citation7,Citation8]. Moreover, activation of the cAMP pathway was shown to suppress inflammatory response, epithelial injury, and fibrosis. Compounds that can modulate inflammatory response, prevent epithelial damage, and protect against PH have potential utility in PH-IPF. We evaluated the efficacy of dual activation of the cAMP/cGMP pathways in the MCT rat model.
In this study, compound A increased both cAMP and cGMP. Profibrotic gene expression induced by TGF-β was attenuated by dual activation of cAMP/cGMP pathways. Activation of cAMP/cGMP pathways by compound A is enough to induce cellular signaling. Elevation of cAMP, as part of the PGE2 pathway, plays an important role in the activation of the anti-fibrotic pathway in lung fibroblasts. Elevated cAMP can phosphorylate cAMP response element binding protein (CREB), which then recruits the coactivators CREB binding protein and p300 to inhibit their function as co-factors for several transcriptional elements, such as small mothers against decapentaplegic (SMAD)3/4, which stimulates profibrotic gene expression [Citation15]. In addition to the cAMP pathway, the cGMP pathway is known to inhibit the profibrotic pathway. Previous studies have shown that sGC activators suppress TGF-β-induced profibrotic pathway, in which elevated cGMP suppresses the nuclear translocation of phospho-SMAD3 from the cytosol by sequestering SMAD3 from beta 2-tubulin [Citation16,Citation17]. The anti-fibrotic effect via dual activation of cAMP/cGMP pathway might thus be mediated via inhibition of the translocation of SMAD3 and interaction between SMAD3 and p300 via activation of cAMP.
Our results also demonstrated the increase in cAMP and cGMP levels by oral administration of compound A in the MCT rat. Moreover, our results showed a clear relationship between compound A plasma concentration, cAMP and cGMP, with a slight delay. In parallel with the elevation in plasma compound A levels, cAMP and cGMP were elevated for 8 h and returned to basal levels at 24 h after a single administration. Our experimental setting for in vivo study using compound A is suitable to evaluate the effect of dual activation of cAMP/cGMP pathways.
To evaluate the effect of dual activation of cAMP/cGMP pathways in modulating pulmonary arterial pressure, we measured RV/LV+S and BNP expression in the right ventricle of MCT rats. RV/LV+S ratio reflects adaptive hypertrophy of the right ventricle and is widely used as an index of elevated pulmonary arterial pressure. BNP gene expression in cardiomyocytes was shown to be elevated by stretch stimulus [Citation18], and its expression level in the right ventricle was shown to reflect pulmonary artery pressure [Citation19]. In this study, compound A reduced RV/LV+S ratio and BNP expression in the right ventricle of MCT rats. Elevation of cGMP in pulmonary arterial smooth muscle cells induces vasodilation by suppression of Ca2+ influx. Protein kinase G, which is activated by cGMP, was shown to suppress the activity of voltage-gated ion channel [Citation20]. Increased Ca2+ ions in the cytoplasm bind to calmodulin and activate myosin light chain kinase and myosin ATPase, leading to cross-binding cycles between myosin and actin filaments [Citation21]. Moreover, increased cGMP can suppress the proliferation of pulmonary arterial smooth muscle cells. Calcium-dependent kinase and calmodulin are involved in the transcription of cell cycle-related genes; thus, depletion of Ca2+ in pulmonary smooth muscle cell inhibits their proliferation [Citation22]. Elevation of cGMP may suppress proliferation of pulmonary arterial smooth muscle cells and the subsequent entry of Ca2+ into the cytoplasm.
Previous studies have demonstrated the presence of inflammatory response and lung injury in MCT rats [Citation23,Citation24]. MCT rat, better known as a model of PH, is also useful for the evaluation of inflammatory response and epithelial injury observed in PH-IPF. Our results demonstrated elevated expression of Ccl2 gene in MCT rats. Ccl2 was shown to be elevated in the lungs of IPF patients [Citation25] and can suppress PGE2 production in epithelial alveolar cells and induce fibroblast proliferation [Citation26]. Although sildenafil did not alter Ccl2 expression, compound A (10 mg/kg) significantly suppressed Ccl2 expression in the lung. Given that compound A at 10 mg/kg elevated cAMP levels in vivo, suppression of Ccl2 may be attributable to the activation of cAMP pathway. A previous study showed that PDE4 inhibitors, which elevate cAMP, suppress Ccl2 production in lung macrophages [Citation27]. Suppression of Ccl2 in the lung by compound A might be due to a reduction in Ccl2 production via activation of cAMP pathway in lung macrophages. SP-D was also reported to be released in blood after epithelial injury. PDE4 inhibitors were shown to suppress apoptosis of epithelial cells [Citation28] by reduction in reactive oxygen species via nicotinamide adenine dinucleotide phosphate oxidase 4. Suppression of Ccl2 and SP-D may be mediated via activation of cAMP pathway in macrophages and epithelial cells, respectively.
Activation of cGMP pathway is a potential treatment option for PH-IPF. Additional activation of cAMP pathway to achieve anti-inflammatory and epithelioprotective effect can be an effective treatment strategy for PH-IPF. Especially, SP-D was shown to be released in blood after epithelial injury and was shown to be elevated in plasma of IPF patients [Citation29]. Sustained injury to epithelial cells can cause IPF [Citation30]. Dual activation of cAMP/cGMP pathways confers beneficial effects not only in pulmonary artery but also in epithelium.
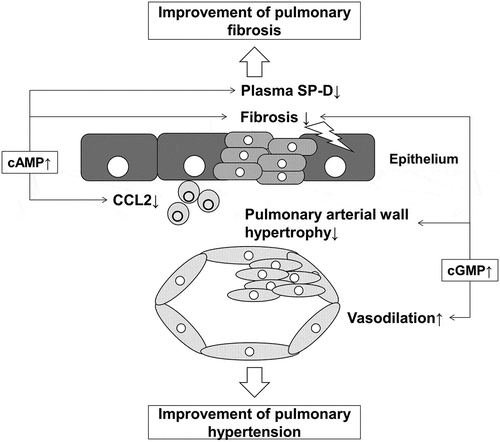
We evaluated the effect of dual activation of cAMP/cGMP pathways on pulmonary hypertension, inflammatory response, and epithelial injury in the MCT rat model of PH. We found that compound A suppressed right ventricular hypertrophy and Ccl2 expression and ameliorated elevated plasma SP-D levels in MCT rats. These effects suggest that dual activation of cAMP/cGMP pathways can modulate the inflammatory response, epithelial injury, and vascular remodeling, which play an important role in the pathogenesis of PH-IPF (). Compound A which can induce dual activation of cAMP/cGMP pathways can be a potential therapeutic candidate for PH-IPF.
Figure 7. Schematic illustration of the mechanism of the potential benefit of dual activation of cAMP/cGMP in PH-IPF.
Elevated cAMP suppresses profibrotic and inflammatory responses. Elevated cGMP enhances vasodilation and suppresses proliferation of smooth muscle cells. These dual effects mediated by cAMP and cGMP can have a potent effect on ameliorating pulmonary hypertension. RV/LV+S, ratio of the right ventricular weight to that of the ventricle plus septum; Ccl2, C─C chemokine 2; SP-D, surfactant protein D.
Author Contribution
Yo Muraki and Takako Naito designed the study protocols, conducted the experiments and analysed data. Kimio Tohyama, Sachio Shibata, Kanako Kuniyeda and Yasunori Nio conducted the experiments and analysed data. Yo Muraki, Masatoshi Hazama and Takanori Matsuo considered research plan, wrote and reviewed the manuscript. All authors discussed the data and reviewed the manuscripts.
Acknowledgments
We are deeply grateful to Sham Nikam (Takeda Pharmaceutical Company Limited) for his supervision and advice for these experiments. We are also grateful to Toshiro Yamashita, Hisashi Anayama, Sachiko Shiotani, and Katsunori Takashima (Takeda Pharmaceutical Company Limited) for their helpful suggestions to advance this project. We appreciate Noriko Uchiyama (Takeda Pharmaceutical Company Limited) for her suggestions for the in vitro study. We also appreciate Masahiro Ide, Jyunichi Taki, and Risa Bunno (TAKEDA RABICS, Ltd) for their support for animal experiments.
Disclosure statement
All authors are employee of Takeda Pharmaceutical Company Limited.
Additional information
Funding
References
- Rosenzweig EB. Emerging treatments for pulmonary arterial hypertension. Expert Opin Emerg Drugs. 2006;11(4):609–619.
- Sardana M, Moll M, Farber HW. Novel investigational therapies for treating pulmonary arterial hypertension. Expert Opin Investig Drugs. 2015;24(12):1571–1596.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34–41.
- Ghofrani HA, D’Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369(4):319–329.
- Collum SD, Amione-Guerra J, Cruz-Solbes AS, et al. Pulmonary hypertension associated with idiopathic pulmonary fibrosis: current and future perspectives. Can Respir J. 2017 20171430350.
- Raghu G, Behr J, Brown KK, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641–649.
- Tanabe N, Taniguchi H, Tsujino I, et al. Multi-institutional retrospective cohort study of patients with severe pulmonary hypertension associated with respiratory diseases. Respirology. 2015;20(5):805–812.
- Han MK, Bach DS, Hagan PG, et al. Sildenafil preserves exercise capacity in patients with idiopathic pulmonary fibrosis and right-sided ventricular dysfunction. Chest. 2013;143(6):1699–1708.
- Ghofrani HA, Wiedemann R, Rose F, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet. 2002;360(9337):895–900.
- Sajkov D, Mupunga B, Bowden JJ, et al. Pulmonary Hypertension in Chronic Lung Diseases and/or Hypoxia, 2013.
- Bozyk PD, Moore BB. Prostaglandin E2 and the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;45(3):445–452.
- Cortijo J, Iranzo A, Milara X, et al. Roflumilast, a phosphodiesterase 4 inhibitor, alleviates bleomycin-induced lung injury. Br J Pharmacol. 2009;156(3):534–544.
- Glickman JF, Schmid A, Ferrand S. Scintillation proximity assays in high-throughput screening. Assay Drug Dev Technol. 2008;6(3):433–455.
- Nio Y, Tanaka M, Hirozane Y, et al. Phosphodiesterase 4 inhibitor and phosphodiesterase 5 inhibitor combination therapy has antifibrotic and anti-inflammatory effects in mdx mice with Duchenne muscular dystrophy. FASEB J. 2017;31(12): 5307–5320.
- Shen X, Hu PP, Liberati NT, et al. TGF-beta-induced phosphorylation of Smad3 regulates its interaction with coactivator p300/CREB-binding protein. Mol Biol Cell. 1998;9(12):3309–3319.
- Gong K, Xing D, Li P, et al. cGMP inhibits TGF-beta signaling by sequestering Smad3 with cytosolic beta2-tubulin in pulmonary artery smooth muscle cells. Mol Endocrinol. 2011;25(10):1794–1803.
- Schinner E, Wetzl V, Schramm A, et al. Inhibition of the TGFbeta signalling pathway by cGMP and cGMP-dependent kinase I in renal fibrosis. FEBS Open Bio. 2017;7(4):550–561.
- Sadoshima J, Jahn L, Takahashi T, et al. Molecular characterization of the stretch-induced adaptation of cultured cardiac cells. An in vitro model of load-induced cardiac hypertrophy. J Biol Chem. 1992;267(15):10551–10560.
- Adachi S, Ito H, Ohta Y, et al. Distribution of mRNAs for natriuretic peptides in RV hypertrophy after pulmonary arterial banding. Am J Physiol. 1995;268(1 Pt 2):H162–169.
- Morgado M, Cairrao E, Santos-Silva AJ, et al. Cyclic nucleotide-dependent relaxation pathways in vascular smooth muscle. Cell Mol Life Sci. 2012;69(2):247–266.
- Kuhr FK, Smith KA, Song MY, et al. New mechanisms of pulmonary arterial hypertension: role of Ca(2)(+) signaling. Am J Physiol Heart Circ Physiol. 2012;302(8):H1546–1562.
- Golovina VA, Platoshyn O, Bailey CL, et al. Upregulated TRP and enhanced capacitative Ca(2+) entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol. 2001;280(2):H746–755.
- Molteni A, Ward WF, Ts’ao CH, et al. Monocrotaline-induced pulmonary fibrosis in rats: amelioration by captopril and penicillamine. Proc Soc Exp Biol Med. 1985;180(1):112–120.
- Ikeda Y, Yonemitsu Y, Kataoka C, et al. Anti-monocyte chemoattractant protein-1 gene therapy attenuates pulmonary hypertension in rats. Am J Physiol Heart Circ Physiol. 2002;283(5):H2021–2028.
- Mercer PF, Johns RH, Scotton CJ, et al. Pulmonary epithelium is a prominent source of proteinase-activated receptor-1-inducible CCL2 in pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179(5):414–425.
- Moore BB, Peters-Golden M, Christensen PJ, et al. Alveolar epithelial cell inhibition of fibroblast proliferation is regulated by MCP-1/CCR2 and mediated by PGE2. Am J Physiol Lung Cell Mol Physiol. 2003;284(2):L342–349.
- Buenestado A, Grassin-Delyle S, Guitard F, et al. Roflumilast inhibits the release of chemokines and TNF-alpha from human lung macrophages stimulated with lipopolysaccharide. Br J Pharmacol. 2012;165(6):1877–1890.
- Milara J, Peiro T, Serrano A, et al. Roflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm Pharmacol Ther. 2014;28(2):138–148.
- Greene KE, King TE Jr., Kuroki Y, et al. Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur Respir J. 2002;19(3):439–446.
- King TE Jr., Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–1961.