
ABSTRACT
Completion of the whole genome sequence of a laboratory yeast strain Saccharomyces cerevisiae in 1996 ushered in the development of genome-wide experimental tools and accelerated subsequent genetic study of S. cerevisiae. The study of sake yeast also shared the benefit of such tools as DNA microarrays, gene disruption-mutant collections, and others. Moreover, whole genome analysis of representative sake yeast strain Kyokai no. 7 was performed in the late 2000s, and enabled comparative genomics between sake yeast and laboratory yeast, resulting in some notable finding for of sake yeast genetics. Development of next-generation DNA sequencing and bioinformatics also drastically changed the field of the genetics, including for sake yeast. Genomics and the genome-wide study of sake yeast have progressed under these circumstances during the last two decades, and are summarized in this article.
Abbreviations: AFLP: amplified fragment length polymorphism; CGH: comparative genomic hybridization; CNV: copy number variation; DMS: dimethyl succinate; DSW: deep sea water; LOH: loss of heterozygosity; NGS: next generation sequencer; QTL: quantitative trait loci; QTN: quantitative trait nucleotide; SAM: S-adenosyl methionine; SNV: single nucleotide variation
Graphical abstract
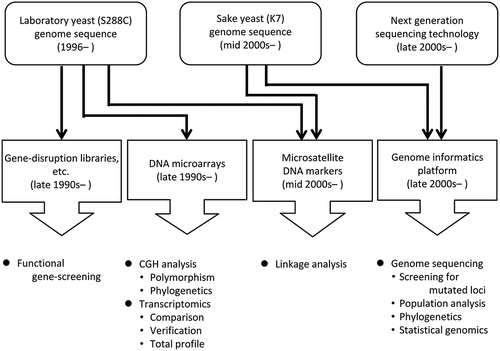
Improvement of genetic infrastructures has supported the recent development of genomics and genome-wide studies of sake yeast.
Although sake yeast strains belong to the Saccharomyces cerevisiae species [Citation1], they possess unique characteristic such as high ethanol productivity, distinguished growth and fermentation at a low temperature under 15ºC, and well-balanced production of flavor compounds that result in a superior organoleptic quality of the final products. Genetic and molecular characterization of such unique characteristics is a major purpose of studies on sake yeast. Unfortunately, most of these characteristics largely remain to be elucidated.
Today, genome-wide and genomic approaches are indispensable for genetic studies. Since the whole genome sequence of the budding yeast Saccharomyces cerevisiae laboratory strain S288C was published in 1996 [Citation2], efficient experimental tools for genome-wide analyses including DNA microarrays [Citation3–Citation5], deletion library of each nonessential gene [Citation6], a collection of GFP-tagged-ORFs [Citation7] were developed in the S288C genetic background ().
Figure 1. Genetic infrastructures and innovative DNA sequencing technologies that supported the recent development of genomics and genome-wide studies of sake yeast.
As described above, the sake and S288C strain both belong to the same species. Hence, in the early stages of the genome-wide study of sake yeast, the tools described above, in particular the DNA microarray and deletion library were employed exclusively, and functioned well as expected. Whole genome information for sake yeast later became available [Citation8]. In addition, next generation sequencing (NGS) technologies were developed and advanced after the late 2000s [Citation9]. Under such circumstances, the study of sake yeast based on genome sequence became common after 2010 (). In this article, genome-wide and genomic studies of sake yeast are reviewed. Firstly, DNA microarray studies of sake yeast will be introduced, and next, whole genome analysis of sake yeast will be summarized, followed by an overview of the recent genome sequence-based studies.
Genome-wide study of sake yeast with DNA microarrays
DNA microarrays are one of the most major experimental tools for genome-wide study. It is a potent hybridization-based experimental tool, and applicable to transcriptomic and comparative genomic studies using cDNA and genomic DNA, respectively. A comparative genomic hybridization (CGH) study between sake yeast Kyokai no. 7 (K7) and X2180, an isogenic diploid of S288C, indicated that the commercially-provided yeast microarray was effectively applicable to the hybridization study of sake yeast although it had been designed based on the S288C genome sequence [Citation10] ().
Characteristic gene expression profiles of sake yeast
Several transcriptomic studies that attempt to explore characteristic features of gene expression of sake yeast have ever been reported. Wu et al. performed transcriptomic analysis of sake yeast during sake fermentation, to determine total gene expression profiles of yeast in sake mash over time [Citation11]. Numerous genes were classified into groups by expression pattern. To date, this has been the only investigation of total gene expression in sake yeast in sake mash, and is referenced by several subsequent studies on the fermentation ability of sake yeast [Citation12–Citation16].
Shobayashi et al. compared total expression profiles of cells in logarithmic phase cultured under shaking and static condition between the sake strain K9 and a laboratory strain X2180-1A [Citation17]. Genes related to carbon metabolism (HXT, ATP, and COX genes), ergosterol biosynthesis (ERG genes), and thiamine metabolism (THI genes) were found to be transcribed at higher level in K9 than in X2180-1A. Meanwhile, CUP1 and PHO genes were transcribed at lower in K9 than X2180, providing an explanation for the low Cu-resistance and the low acid phosphatase activity in K9 [Citation17].
Another comparative study between K7 and X2180-1A found that the expression level of the Δ9-fatty acid desaturase gene OLE1 and the oleic acid content in the plasma membrane were related to the fermentation ability and ethanol tolerance of yeast cells [Citation18,Citation19]. Transcriptomic analysis using GeneFilters [Citation5] a membrane-based DNA microarray, revealed that the Δ9-fatty acid desaturase gene OLE1 was expressed at higher level in K7 than in the laboratory strain. This result was consistent with the higher content of unsaturated fatty acids in K7. Furthermore, overexpression of OLE1 in the laboratory strain resulted in unsaturated fatty acid levels similar to sake yeast. High expression levels of OLE1 were commonly observed in other sake yeast strains such as K9 and K10, resulting in increased levels of unsaturated fatty acids in sake yeast cells [Citation20].
Ergosterol is well known to be a component related to the maintenance of membrane integrity and ethanol tolerance in yeast cells [Citation21,Citation22]. Tamura et al. revealed with a microarray experiment that the ergosterol biosynthetic pathway genes ERGs and their upstream regulator gene HMG1 were transcribed at higher levels in K7 than in X2180 with the microarray experiment. The difference in the expression levels of the genes was naturally reflected in the ergosterol content of the cells. X2180 HAP1 was found to be mutated by the insertion of a Ty1 long terminal repeat around its C-terminal region. Complementation of the K7 HAP1 allele in X2180 disclosed that the aberrant HAP1 allele caused the decreased expression of ERG and HMG1 genes in X2180 cells [Citation23]. Similarly, a difference in the transcription levels of the ERG genes between sake yeast strain K9 and a laboratory strain X2180-1A was observed in another transcriptome analysis (in K9 > in X2180-1A) [Citation17,Citation24].
To screen for genes involved in ethanol tolerance, Hirasawa et al. analyzed and compared gene expression profiles between K7 and a laboratory strain FY834 under conditions with and without 5% ethanol in the medium [Citation24]. They found elevated expression of tryptophan-biosynthetic genes in FY834 with 5% ethanol, and confirmed that tryptophan has an effect on the enhancement of ethanol tolerance in a laboratory strain.
Assessment of mutant physiology through the gene expression profile
In the sake brewing industry, mutant strains that have acquired some characteristic features are favored. Thus many mutated strains have ever been developed and practically used in brewing industry. Transcriptomic analyses have frequently been employed to better understand the physiological state of such mutated cells through observation of gene expression (a)).
Figure 2. Different approaches for better understanding physiology of the mutant cell and more optimized industrial use. (a) Approach from transcriptome analysis using DNA microarrays, (b) Approach from comparative genomics.
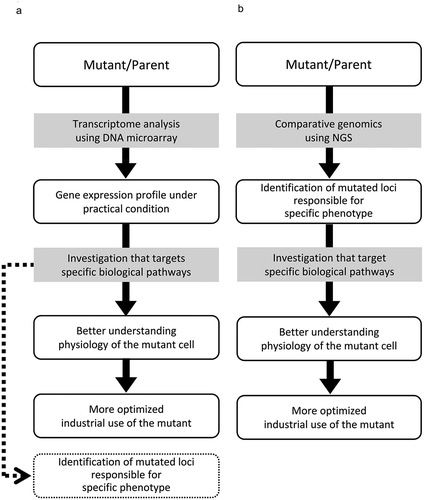
Two alcohol tolerant mutants derived from K7, SR4-3 [Citation25] and K11 [Citation26], were independently applied to transcriptome analysis. Both of them showed elevated expression of a series of stress-responsive genes, which possessed the stress-responsive cis-element STRE in their promoter regions, and were regulated by the DNA-binding transcription factor Msn2/4p [Citation27–Citation30]. Since SR4-3 and K11 also possess similar properties such as high malic acid production and killer toxin resistance [Citation26,Citation28–Citation30], an intriguing question is whether their responsible genes are identical to each other.
In an α-ketoglutarate-resistant sake yeast mutant strain with high organic acid productivity [Citation31,Citation32], transcriptome analysis revealed that genes involved in TCA cycle, oxidative phosphorylation, and respiration were expressed at higher levels than the parental strain. These genes are transcriptionally regulated by the Hap2/3/4/5p complex [Citation33]. In a subsequent study, elevated expression of HAP4 was observed in the mutant, and overexpression of HAP4 in the parent caused enhanced production of the organic acids [Citation32].
In a K9-dereived strain with high malic acid productivity that was isolated as natural variant, thiamine-biosynthetic genes were depressed. This implied that a shortage of thiamine caused high malic acid synthesis through accumulation of pyruvic acid [Citation34]. In another high malic acid producing strain with cycloheximide-resistance, derived from K7, transcriptional levels were elevated for genes MDH1 and MDH2, both encoding malate dehydrogenase, and ICL1 encoding isocitrate lyase [Citation35].
It has been well known that industrial sake yeast cells can produce and accumulate high level of S-adenosyl methionine (SAM) [Citation36]. Kanai et al. functionally screened 123 methionine-resistant strains from the single gene deletion series of the laboratory strain BY4742, to developing a more efficient method for producing SAM in yeast. Among the 123 strains tested, the Δado1 strain was revealed to accumulate SAM most efficiently. Results of transcriptome analysis and other analysis of the Δado1 strain suggested that activation of the methionine-biosynthetic pathway caused SAM accumulation in the strain [Citation36,Citation37]. Based on these results, an efficient method to screen SAM accumulating yeast was developed, in which cordycepin resistance was employed as a selective marker [Citation38].
Other than the studies described above, transcriptomic analysis was carried out and reported for several mutant strains that showed specific phenotypes including: elevated production of flavor components such as isoamyl acetate [Citation39,Citation40] or β-phenylethyl alcohol [Citation41]; resistance to chemical drugs such as 2-deoxyglucose [Citation42], hygromycin B [Citation39], trifluoroleucine [Citation40], or trichothecin [Citation43].
Verification of gene expression profiles needed for trouble-free industrial use
In some situations, DNA microarrays were also used to verify the yeast gene expression profile and determine whether some serious defects in the transcriptome emerged and led to disturbances in fermentation. Here, three examples are given below.
Uehigashi et al. examined the effects of deep sea water (DSW), collected from 320 meters below sea level in the Muroto area of Kochi Prefecture, on fermentation by sake yeast. Transcriptome analysis indicated that addition of DSW to fermentation caused elevated transcription of genes involved in the biosynthesis of higher alcohols and fatty acids, resulting in facilitation of favorable volatile compounds such as isoamyl acetate, ethyl caproate, and ethyl caprylate. Intriguingly, whereas addition of just NaCl led to an increase in stress-responsive gene expression, addition of DSW including the same level of NaCl did not [Citation44].
Dried and wet sake yeast samples were transferred to the International Space Station by the Russian spaceship Soyuz, kept there for ten days, and then brought back to earth. Transcriptome analysis was performed to evaluate the effect on yeast of staying in the space station, and revealed that the expression profiles of the dried samples were almost identical to the control samples maintained on earth. On the other hand, although the expression profiles of the wet samples were different to the controls, serious defect of the strain was not observed in sake brewing characteristics [Citation45].
Ethyl carbamate is an unfavorable component of sake, which possibly shows weak carcinogenic activity in mammals, and is generated thorough a nonenzymatic reaction between urea and ethanol in sake products [Citation46]. In Canada, self-cloned sake yeast strains were developed to minimize ethyl carbamate formation in sake products. DUR1/DUR2 and DUR3 genes encoding urea amidolyase and urea permease, respectively, were introduced into K7 and K9 and designed to be constitutively co-expressed. A DNA microarray experiment was employed to indicate that these strains were nearly equivalent to the parental strains from a transcriptional perspective [Citation47]. Nevertheless, as self-cloned strains, they have not been approved for use in industrial sake production under Japanese regulations. In addition, an efficient culture medium for selection of urea non-producing yeast mutants has already been developed [Citation48].
Distinctiveness of sake yeast genome compared with laboratory yeast
Although the unique features of sake yeast have been recognized for a long time, the genetic background of such characteristics remains disappointingly veiled. The whole genome sequence of laboratory yeast S288C was released in the mid 1990s [Citation1], and provided a major turning point for the yeast research including sake yeast. Over time, multiple studies clarified the differences in genomic characteristics between the sake and laboratory strains.
Nearly all the wild-type strains of sake yeast generate a foam layer on the mash surface during fermentation, differentiating them from the laboratory yeast and the other industrial strains. Accordingly, foamless mutants have been bred from various industrial sake yeast strains and widely used in industrial sake production [Citation49,Citation50]. Soon afterward a gene responsible for foam layer formation in sake mash was characterized in the K7 genome and named AWA1 (“awa” is a Japanese term for foam). The AWA1 ORF has a unique structure, like a chimera between two identified S288C genes DAN4/YJR151C and HPF1/YOL155C [Citation51].
Sake yeast strains possess the ability to synthesize biotin differently from S288C. The K7 genome was partially sequenced with the shot gun method [Citation52], which led to the characterization of the novel gene BIO6. The gene was revealed to be essential for biotin biosynthesis in S. cerevisiae cells, and its distribution within the species was mostly limited to sake yeast strains [Citation53]. The discovery of AWA1 and BIO6 were the earliest examples of differentially distributed genes among S. cerevisiae strains.
CGH analysis is another application of DNA microarrays study other than the transcriptome. CGH analysis of K7 and S288C detected copy number variations (CNVs) in the K7 genome. In particular, K7 possesses additional two copies of an approximately 22kb-sequence corresponding to the AQY1–ARR3 region of chromosome XVI in the K7 and S288C genomes. The additional copies in the K7 genome were located in the next-to-telomeric-repeat regions of chromosomes IV and XIII, suggesting that amplification of the region in the K7 genome might have resulted from nonreciprocal translocation events by homologous recombination via telomeric repeats [Citation10] (). In addition, preliminary sequencing of the K7 genome also identified several structural and numerous microsatellite polymorphisms between the K7 and S288C genomes [Citation52]. The presence of the differential genes, CNVs, and microsatellite polymorphisms clearly showed the genomic diversity within S. cerevisiae species.
Meanwhile, an amplified fragment length polymorphism (AFLP) analysis indicated that commonly used sake yeast strains such as K6, K7, K9, K10 and their derivatives, which are collectively called the K7 group for convenience in classification, were indistinguishable with this method although they were clearly differentiated from other industrial yeast [Citation54]. Consequently, genome information from representatives of these strains strongly contributed to genetic and genomic understanding of common sake yeast strains such as the K7 group. It is likely that the situation at that time (first half of the 2000s) raised momentum for the whole genome analysis of sake yeast, which is described later.
Linkage analyses using genome-wide DNA markers
As stated above, microsatellite polymorphisms were identified between the K7 and S288C genomes, followed by development of genome-wide microsatellite DNA markers [Citation52]. This set of DNA markers was applied to linkage analysis for chromosomal mapping of characteristic genetic traits of sake yeast [Citation55,Citation56] (). Many industrially important features of sake yeast are unique to sake yeast strains and are not observed in laboratory strains [Citation8]. These features of sake yeast include initial fermentation rate, final ethanol concentration, production of an appropriate amount of organic acid and volatile components, etc. These are known to be quantitative traits, for which the phenotype is determined by the genotypes of multiple loci [Citation55,Citation57]. Therefore, Kato et al. performed quantitative trait loci (QTL) analysis, a type of the linkage analysis, to characterize these traits. A K7 haploid (MATa) was mated to X2180-1B (MATα) to produce a hybrid diploid strain. A hundred of haploid segregants were isolated from the hybrid, and then subjected to a small-scale sake brewing test to collect phenotypic data concerning sake brewing. Subsequently, QTL analysis was carried out for the segregants using 142 microsatellite DNA markers with heterogeneity between the two parental strains. As a result, 25 significant QTLs that were involved in ethanol fermentation and the production of volatiles were successfully identified. Unfortunately, identification of the quantitative trait nucleotides (QTNs) responsible for such characteristic features was not achieved [Citation55].
Kanai et al. used the same set of segregants and markers as Kato et al. [Citation52,Citation55], and explored QTLs responsible for the higher productivity of SAM in K7 [Citation36,Citation37,Citation56]. This study identified two significant QTLs, and one became the focus of further investigation. Among a total of 165 genes located in the 218.5-kb QTL region, ERC1 was suspected to be a responsible gene due to its gene function, encoding a member of the multi-drug and toxin extrusion family, draw attention as a responsible gene by considering gene function. In addition, by referring to the K7 genome information [Citation8], a single-nucleotide variation (SNV) was found in ERC1, where single adenine nucleotide was deleted in K7 (K7ERC11628delA), and finally identifying ERC1 as a QTN responsible for the increased SAM accumulation in K7 cells [Citation56]. At present, this is the only successfully identified QTN from sake yeast linkage analysis.
Whole genome analysis of the strain K7 and its impact on sake yeast genetics
A whole genome sequencing project of sake yeast was conducted thorough a collaboration between industry and academia [Citation8,Citation58]. As a source of genomic DNA, the project chose one of the most major strains, K7. Whole genome shotgun sequencing was done by the Sanger method, followed by de novo assembly with the Phred-Phrap-Consed package [Citation59]. Comparative study of the K7 and S288C genomes revealed a large number of features in the K7 genome (). The assembled sequence of K7 was nearly identical to that of S288C, although several subtelomeric polymorphisms and internal inversions on chromosome V and XIV were observed in K7. A genome-wide survey of heterozygous sites revealed an uneven distribution of heterozygosity through the genome. Sequential loss of heterozygosity (LOH) events would have resulted in such a mosaic-like distribution pattern, suggesting that LOH was one of the major driving forces in genetic diversification of sake yeast, as well as other monocellular diploidic microorganisms. Many gene differences were identified between the two strains: a total of 48 K7 genes were absent in S288C, and 49 S288C genes were absent in K7, most of which were located in subtelomeric regions of the chromosomes. Amino acid sequences of a large part of the K7 ORFs were almost identical (>98% on average) to S288C orthologous ORFs. However, structural polymorphisms such as those caused by frameshift or nonsense mutations that might cause loss- or change-of-function mutations were discovered in ORFs of approximately 200 orthologous pairs of K7 and S288C. Distribution of Ty elements was also quite different between them K7 and S288C ().
Table 1. Characteristic features found in the K7 genome structure, compared to the S288C genome.
This study can be regarded as a landmark one for genetics of sake yeast. Subsequent studies were supported by the genome information and identified loss-of-function mutations in the K7 allele of genes indicated to be involved in alcohol fermentation: msn4C1540T, rim155055insA (rim155067ins A based on position in the K7 allele), Δppt1::Ty2, and glg2G1086A [Citation13,Citation15,Citation16,Citation60,Citation61]. Moreover mutations except for glg2G1086A, were confirmed to be distributed commonly among other strains of the K7 group [Citation13,Citation58,Citation60], and to be available as DNA markers to discriminate the K7-group strains from other S. cerevisiae strains [Citation62]. These results considerably improved our understanding of regulation of alcohol fermentation in yeast cells [Citation13,Citation15,Citation16,Citation60,Citation61].
Genomic studies with NGS
Screening for responsible polymorphic sites of sake yeast strains with specific characteristics
Transcriptome analysis of mutant strains has been favored to evaluate the physiological state of mutant cells through the total gene expression profile of the cells (). Meanwhile, recent progress in the comparative genomic approach by use of the NGS has brought about changes in the analysis of the mutant strains. In such studies, to elucidate mutant cell physiology for advanced use of the mutant, successful identification of the responsible gene and polymorphic site for specific characteristics of mutant cells was followed by targeted investigation of some sort of intracellular signal transduction and/or metabolic pathways ()). Several studies were carried out to identify mutated loci responsible for specific characteristics of the mutant strains () [Citation63–Citation66]. There, the K7 genome was preferred as the reference sequence ().
High malic acid-productive mutants of sake yeast, F-701H and K-901H were bred by use of sensitivity to dimethyl succinate (DMS), a specific inhibitor of succinate dehydrogenase, as a phenotypic marker [Citation63,Citation67]. Negoro et al. identified a missense mutation in the VID24 gene, which is a member of the glucose-induced-degradation-deficient (GID) complex, in K-901H cells by comparative genomics, and revealed that the mutation, VID24G391A, is responsible for high malic acid production and DMS sensitivity of the cells. They found that Mdh2p, a malate dehydrogenase isozyme, is involved with high malic acid production in the cells with the VID24G319A mutation, and observed accumulation of Mdh2p in vid24-disrupted yeast cells [Citation63].
Negoro et al. also identified the nonsense mutation pex22374A as responsible for the high malic acid productivity of F-701H by using the same approach as above. The mutant and parental strains harbored the mutation homozygously and heterozygously, respectively. The PEX22 gene encodes one of the peroxin family proteins, in which are involved in peroxisome biogenesis and transport of proteins with a peroxisomal targeting signal 1 (PTS1) to the peroxisomal matrix. A malate dehydrogenase isozyme, Mdh3p, is one of such proteins along with PTS1. In the mutant cells, a change in Mdh3p localization from the peroxisome to the cytosol was observed, implying that mislocalization of Mdh3p caused high malate production [Citation64].
Takahashi et al. isolated sake yeast strains that produce high levels of isoamyl acetate, from mutants resistant to aureobasidin A, which is an inhibitor of the inositol phosphorylceramide synthase. The mutant strains hia1, hia2, hia4, and hia6 showed elevated transcription of the ATF1 gene encoding alcohol acetyltransferase, and were able to produce sake product containing high levels of isoamyl acetate even if lowly polished rice was used as raw material. Pooled genome sequencing analysis of the four hia mutants was performed, and it was revealed that a homozygous nonsense mutation (Ser706*) within MGA2, a gene for a transcription factor of ATF1, was responsible for the high isoamyl acetate production of the mutants [Citation65].
Takahashi et al. also isolated three mutants (hec2, hec3, and hec6) from hia1 using cerulenin-resistance as a selective marker. The hec mutants exhibited elevated caproate production during sake brewing, in addition to isoamyl acetate. Genome sequencing of hec2 revealed a mutation in the FAS1 gene (FAS1G2725C), which encodes a subunit of fatty acid synthase in addition to FAS2G3748A, a known polymorphism causing high level of ethyl caproate production [Citation68]. In addition, it was found that transcription level of EEB1, a gene for alcohol acyltransferase, was elevated, and that the mutant allele of MGA2 in hia1 and hec2 facilitated transcription of FAS1and FAS2. Since the high ethyl caproate productivity of hec2 could not be explained solely by mutant FAS2 alleles, it was inferred to be caused by the compound effect of enhanced EEB1 transcription, and mutant alleles of MGA2, FAS1 and FAS2 [Citation66].
High pyruvate concentration during sake storage results in the generation of off-flavor compounds such as diacetyl and acetaldehyde. Low pyruvate producing yeast was isolated from strains resistant to ethyl-α-transcyanocinnamate, an inhibitor of mitochondrial pyruvate transport [Citation69]. Kadowaki et al. conducted genome sequencing analysis of the mutated strain and reported that some sort of combination of chromosomal aneuploidies, led to the pyruvate productivity of the mutated strain [Citation70].
Aside from the studies above, the genomes of various sake and related yeast strains has been sequenced with NGS. The obtained read data were mapped to the reference genomes of K7 and S288C [Citation2,Citation8], and polymorphic sites for each strain were identified (unpublished data). Comparative genomic analysis for a large number of such yeast strains led to the discovery of numerous functional mutations in the CDC55 gene. These mutations were reported to be involved in the maintenance of cell-morphological robustness [Citation71–Citation73] and regulation of alcohol fermentation [Citation74].
Relatively frequent aneuploidy: novel aspects of the sake yeast strains revealed by NGS studies
Over the past several years, chromosomal aneuploidy has become recognized as one of key factors when considering the relationship between genomic features and phenotypic features. In general, chromosomal aneuploidy confers extensive CNV, and leads to quantitative transcriptomic and proteomic changes. It can cause phenotypic variations in the yeast cells, and contribute to adaptive evolution [Citation75,Citation76]. A recent large-scale genome sequencing study of S. cerevisiae revealed that gain and loss of aneuploidy were observed in the genomes of sake yeast more frequently than in the genomes of other yeast strains [Citation77].
As described above, chromosomal aneuploidy was detected in the mutant strains of sake yeast [Citation70]. In addition, different states of aneuploidy were observed in strains belonging to a single lineage of sake yeast strains (unpublished data). These results imply that the gain and loss of aneuploidy may occur during vegetative growth spontaneously or artificially, although the influence of aneuploidy on brewing characteristics of individual cells or populations is unknown. The frequency and mechanism of the gain and loss of aneuploidy within sake yeast populations are also intriguing questions that remain to be resolved.
As a different matter, aneuploidy was observed in haploids of sake yeast with very high frequency [Citation55,Citation70,Citation78; unpublished data]. Industrial sake yeast strains have almost lost normal spore formation ability [Citation79]. Under spore formation conditions, spores rarely form and the viability of the spores is very low. This deficiency largely prevents cross breeding of sake yeast. Interestingly, a previous study revealed that large proportion of the rarely obtained haploid strains were aneuploid, implying defects in chromosome distribution caused by defective meiotic recombination [Citation55; unpublished data]. Shimoi et al. demonstrated that the strain K7 harbored meiotic recombination deficiency, by genome sequencing of multiple haploid strains and investigating the segregation pattern of two heterozygous sites on the same chromosomes [Citation78]. If the mutations responsible for the defect were identified, genetic characterization of valuable features of sake yeast might easily be achieved by linkage analysis using genetically complemented strains, and cross breeding of new sake yeast strains could be possible.
Genomic studies of shochu yeast
Shochu is a traditional Japanese distilled spirit, which is mainly manufactured in the South Kyushu District and the Izu Islands. Along with sake yeast, the current picture of shochu yeast genomics is summarized here. Shochu yeast strains genetically resemble sake yeast and belong to a “sake clade” within S. cerevisiae species. Although the genetic study of shochu yeast lags behind of that of sake yeast, it has recently been progressing at rapid speed. The strain Kagoshima no. 2 is one of the most popular shochu strains, and its whole genome sequence was obtained through multiple NGS platforms [Citation80], Whole genome sequence was also obtained for the private strain BAW-6, suited for barley shochu fermentation [Citation81]. Interestingly, a nonsense mutation was present in the RIM15 gene of Kagoshima no. 2. A loss-of-function mutation identified in RIM15 was found to be involved in high fermentation ability in common sake yeast strains such as K7 [Citation60]. While the position and type of mutations were different between the common sake yeast strains and the strain Kagoshima no. 2, the loss-of-function mutation in RIM15 would also have a beneficial effect on fermentation ability in Kagoshima no. 2 [Citation80].
Futagami et al. screened for conserved coding sequences among six yeast strains with published whole genome sequences, and selected five genes (THI7, ZAP1, PXL1, GLG1, and YPR1) as suitable loci to elucidate the genome-level phylogeny of the tested strains. They subsequently carried out phylogenetic analysis of shochu, putative indigenous shochu, and 24 previously classified strains using partial sequence of the marker genes, and they found that the shochu related strains formed a monophyletic group as a sub-clade within the sake clade [Citation82].
Future prospects
Today, genomics and genome-wide study of sake yeast have just begun. Genetic mapping of almost every valuable trait remains to be clarified. Comprehensive genomic sequencing of sake and related strains, which is ongoing, will reveal additional novel aspects of the sake yeast genome, and will provide information on nucleotide polymorphisms that will result in high-resolution SNV markers indispensable for more advanced lineage discrimination, linkage analysis, and statistical genetics (). In particular, traditional genetics such as linkage analysis should help us better understand the nature of sake yeast, along with restoration of the meiotic chromosomal recombination defect in sake yeast, which is needed for normal sporulation [Citation78]. In addition, RNA sequencing, one of the NGS applications is a newer approach to the transcriptome, and often employed in recent transcriptomic study [Citation9]. Although there is no reported RNA sequencing study of sake yeast, it may replace DNA microarray in the field of sake yeast research in the near future, resulting in advanced understanding of sake yeast transcriptomics.
Disclosure statement
No potential conflict of interest was reported by the author.
References
- Vaughan-Martini A, Martini A. Saccharomyces Meyen ex Reess (1870). In: Kurtsman CP, Fell JW, Boekhaut T, editors. The yeasts, a taxonomic study. 5th ed. London: Elsevier; 2011. p. 733–746.
- Goffeau A, Barrell BG, Bussey H, et al. Life with 6000 genes. Science. 1996;274(5287):546–567.
- Lashkari DA, DeRisi JL, McCusker JH, et al. Yeast microarrays for genome wide parallel genetic and gene expression analysis. Proc Natl Acad Sci USA. 1997;94(24):13057–13062.
- Johnston M. Gene chips: array of hope for understanding gene regulation. Curr Biol. 1998;8(5):R171–174.
- Cox KH, Pinchak AB, Cooper TG. Genomewide transcriptional analysis in S. cerevisiae by mini-array membrane hybridization. Yeast. 1999;15(8):708–713.
- Winzeler EA, Shoemaker DD, Astromoff A, et al.. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285(5429):901–906.
- Huh WK, Falvo JV, Gerke LC, et al. Global analysis of protein localization in budding yeast. Nature. 2003;425(6959):686–691.
- Akao T, Yashiro I, Hosoyama A, et al. Whole-genome sequencing of sake yeast Saccharomyces cerevisiae Kyokai no. 7. DNA Res. 2011;18(6):423–434.
- Levy SE, Myers RM. Advancements in next-generation sequencing. Annu Rev Genomics Hum Genet. 2016;17:95–115.
- Ogihara F, Kitagaki H, Wang Q, et al. Common industrial sake yeast strains have three copies of the AQY1–ARR3 region of chromosome XVI in their genomes. Yeast. 2008;25(6):419–432.
- Wu H, Zheng X, Araki Y, et al. Global gene expression analysis of yeast cells during sake brewing. Appl Environ Microbiol. 2006;72(11):7353–7358.
- Wu H, Watanabe T, Araki Y, et al. Disruption of ubiquitin-related genes in laboratory yeast strains enhances ethanol production during sake brewing. J Biosci Bioeng. 2009;107(6):636–640.
- Watanabe D, Wu H, Noguchi C, et al. Enhancement of the initial rate of ethanol fermentation due to dysfunction of yeast stress response components Msn2p and/or Msn4p. Appl Environ Microbiol. 2011;77(3):934–941.
- Urbanczyk H, Noguchi C, Wu H, et al. Sake yeast strains have difficulty in entering a quiescent state after cell growth cessation. J Biosci Bioeng. 2011;112(1):44–48.
- Noguchi C, Watanabe D, Akao T, et al. Constitutive hyperphosphorylation of Hsf1p associated with defective ethanol stress response in sake yeast. Appl Environ Microbiol. 2012;78(2):385–392.
- Watanabe D, Hashimoto N, Mizuno M, et al. Accelerated alcoholic fermentation caused by defective gene Expression related to glucose derepression in Saccharomyces cerevisiae. Biosci Biotechnol Biochem. 2013;77(11):2255–2262.
- Shobayashi M, Ukena E, Fujii T, et al. Genome-wide expression profile of sake brewing yeast under shaking and static conditions. Biosci Biotechnol Biochem. 2007;71(2):323–335.
- Kajiwara S, Aritomi T, Suga K, et al. Overexpression of the OLE1 gene enhances ethanol fermentation by Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2000;53(5):568–574.
- You KM, Rosenfield CL, Knipple DC. Ethanol tolerance in the yeast Saccharomyces cerevisiae is dependent on cellular oleic acid content. Appl Environ Microbiol. 2003;69(3):1499–1503.
- Yamada T, Shimoi H, Ito K. High expression of unsaturated fatty acid synthesis gene OLE1 in sake yeasts. J Biosci Bioeng. 2005;99(5):512–516.
- Daum G, Lees ND, Bard M, et al. Biochemistry, cell biology and molecular biology of lipids of Saccharomyces cerevisiae. Yeast. 1998;14(16):1471–1510.
- Inoue T, Iefuji H, Fujii T, et al. Cloning and characterization of a gene complementing the mutation of an ethanol-sensitive mutant of sake yeast. Biosci Biotechnol Biochem. 2000;64(2):229–236.
- Tamura K, Gu Y, Wang Q, et al. A hap1 mutation in a laboratory strain of Saccharomyces cerevisiae results in decreased expression of ergosterol-related genes and cellular ergosterol content compared to sake yeast. J Biosci Bioeng. 2004;98(3):159–166.
- Hirasawa T, Yoshikawa K, Nakakura Y, et al. Identification of target genes conferring ethanol stress tolerance to Saccharomyces cerevisiae based on DNA microarray data analysis. J Biotechnol. 2007;131(1):34–44.
- Nitta A, Uchiyama H, Imamura T. Breeding of ethanol-tolerant sake yeasts from K1 Killer-resistant mutants. Seibutu Kougaku. 2000;78(3):77–81. (in Japanese).
- Hara S, Nojiro K. A remarkably high alcohol-tolerant mutant: application to sake brwing (2). J Brew Soc Jpn. 1978;73(2):76–79. (in Japanese).
- Kobayashi N, McEntee K. Identification of cis and trans components of a novel heat shock stress regulatory pathway in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13(1):248–256.
- Ogawa Y, Nitta A, Uchiyama H, et al. Tolerance mechanism of the ethanol-tolerant mutant of sake yeast. J Biosci Bioeng. 2000;90(3):313–320.
- Ogawa Y. Characters of the ethanol-tolerant mutant of sake yeast. J Brew Soc Jpn. 2001;96(11):730–736. (in Japanese).
- Watanabe M, Tamura K, Magbanua JP, et al. Elevated expression of genes under the control of stress response element (STRE) and Msn2p in an ethanol tolerance sake yeast Kyokai no.11. J Biosci Bioeng. 2007;104(3):163–170.
- Asano T, Yano S, Takakura Y, et al. Isolation of strains with high malate and succinate productivity from α-ketoglutarate resistant mutants of sake yeast. J Brew Soc Jpn. 2003;98(3):217–220. (in Japanese).
- Yano S, Asano T, Kurose N, et al. Characterization of an alpha-ketoglutarate-resistant sake yeast mutant with high organic acid productivity. J Biosci Bioeng. 2003;96(4):332–336.
- Olesen J, Hahn S, Guarente L. Yeast HAP2 and HAP3 activators both bind to the CYC1 upstream activation site, UAS2, in an interdependent manner. Cell. 1987;51(6):953–961.
- Oba T, Suenaga H, Nakayama S, et al. Properties of a high malic acid-producing strains of Saccharomyces cerevisiae isolated from sake mash. Biosci Biotechnol Biochem. 2011;75(10):2025–2029.
- Koganemaru K, Kanda K, Yasuda M, et al. Gene expression concerning malate production of a cycloheximide-resistant mutant and industrial scale sake brewing. J Brew Soc Jpn. 2003;98(4):303–309. (in Japanese).
- Kanai M, Mizunuma M, Fujii T, et al. A genetic method to enhance the accumulation of S-adenosylmethionine in yeast. Appl Microbiol Biotechnol. 2017;101(4):1351–1357.
- Kanai M, Masuda M, Takaoka Y, et al. Adenosine kinase-deficient mutant of Saccharomyces cerevisiae accumulates S-adenosylmethionine because of an enhanced methionine biosynthesis pathway. Appl Microbiol Biotechnol. 2013;97(3):1183–1190.
- Iefuji H, Yasuda N, Kanai M, et al. Method for breeding yeast strain highly accumulating S-adenosylmethionine. Japan Patent P641192. 2014 Nov 7 (in Japanese).
- Inoue T, Iefuji H, Katsumata H. Characterization and isolation of mutants producing increased amounts of isoamyl acetate derived from hygromycin B-resistant sake yeast. Biosci Biotechnol Biochem. 2012;76(1):60–66.
- Oba T, Yamamoto Y, Nomiyama S, et al. Properties of a trifluoroleucine-resistant mutant of Saccharomyces cerevisiae. Biosci Biotechnol Biochem. 2006;70(7):1776–1779.
- Koganemaru K, Sumi T, Kanda K, et al. Beta-phenylethyl alcohol production by a mutant of sake yeast and its highly productive mechanism. J Brew Soc Jpn. 2003;93(3):201–209. (in Japanese).
- Tsuboi H, Wakisaka Y, Hirotsune M, et al. Analysis of the pyruvate permease gene (JEN1) in glucose derepression yeast (Saccharomyces cerevisiae) isolated from a 2-deoxyglucose-tolerant mutant, and its application to sake making. Biosci Biotechnol Biochem. 2003;67(4):765–771.
- Koganemaru K, Sumi T, Kanda K, et al. Improvement of the ethyl alcohol production ability of high malic acid sake yeast. J Brew Soc Jpn. 2004;99(5):365–373. (in Japanese).
- Uehigashi H, Kato R, Sugiyama H, et al. Effect of deep seawater on sake yeast. J Brew Soc Jpn. 2006;101(2):117–124. (in Japanese).
- Kato R, Uehigashi H. Study on making high-quality sake using DNA microarray analysis. The Report of the Works on Kochi Prefectural Industrial Technology Center. 2007; 38:1–7. (in Japanese).
- Alexander J, Auðunsson GA, Benford D, et al. Ethyl carbamate and hydrocyanic acid in food and beverages1Scientific opinion of the panel on contaminants. EFSA J. 2007;551:1–41.
- Dahabieh MS, Husnik JI, Van Vuuren HJ. Functional enhancement of sake yeast strains to minimize the production of ethyl carbamate in sake wine. J Appl Microbiol. 2010;109(3):963–973.
- Kitamoto K, Oda-Miyazaki K, Gomi K, et al. Mutant isolation of non-urea producing sake yeast by positive selection. J Ferment Bioeng. 1993;75(5):359–363.
- Ouchi K, Akiyama H. Non-foaming mutants of sake yeasts selection by cell agglutination method and by froth flotation method. Agr Biol Chem. 1971;35(7):1024–1032.
- Ouchi K. History of non foaming yeasts. J Brew Soc Jpn. 2010;105(4):184–187. (in Japanese).
- Shimoi H, Sakamoto K, Okuda M, et al. The AWA1 gene is required for the foam-forming phenotype and cell surface hydrophobicity of sake yeast. Appl Environ Microbiol. 2002;68(4):2018–2025.
- Namise M, Hata Y, Shimoi H, et al. Genome-wide DNA marker of yeast Saccharomcyes cerevisiae. Japan Patent P2007-82431A. 2005 Sep 20. (in Japanese).
- Wu H, Ito K, Shimoi H. Identification and characterization of a novel biotin biosynthesis gene in Saccharomyces cerevisiae. Appl Environ Microbiol. 2005;71(11):6845–6855.
- Azumi M, Goto-Yamamoto N. AFLP analysis of type strains and laboratory and industrial strains of Saccharomyces sensu stricto and its application to phenetic clustering. Yeast. 2001;18(12):1145–1154.
- Katou T, Namise M, Kitagaki H, et al. QTL mapping of sake brewing characteristics of yeast. J Biosci Bioeng. 2009;107(4):383–393.
- Kanai M, Kawata T, Yoshida Y, et al. Sake yeast YHR032W/ERC1 haplotype contributes to high S-adenosylmethionine accumulation in sake yeast strains. J Biosci Bioeng. 2017;123(1):8–14.
- Katou T, Kitagaki H, Akao T, et al. Brewing characteristics of haploid strains isolated from sake yeast Kyokai No. 7. Yeast. 2008;25(11):799–807.
- Akao T. The genome analysis of sake yeast Kyokai no. 7 and foresights of sake yeast genomics. J Brew Soc Jpn. 2012;107(6):366–380. (in Japanese).
- Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8(3):195–202.
- Watanabe D, Araki Y, Zhou Y, et al. A loss-of-function mutation in the PAS kinase Rim15p is related to defective quiescence entry and high fermentation rates in Saccharomyces cerevisiae sake yeast strains. Appl Environ Microbiol. 2012;78(11):4008–4016.
- Watanabe D, Zhou Y, Hirata A. Inhibitory role of Greatwall-like protein kinase Rim15p in alcoholic fermentation via upregulating the UDP-glucose synthesis pathway in Saccharomyces cerevisiae. Appl Environ Microbiol. 2016;82(1):340–351.
- Akao T, Zhou Y, Watanabe D, et al. Development of DNA markers to differntiate good Kyokai sake yeast and other yeast strains. J Brew Soc Jpn. 2018;113(10):631–641. (in Japanese).
- Negoro H, Kotaka A, Matsumura K, et al. Enhancement of malate-production and increase in sensitivity to dimethyl succinate by mutation of the VID24 gene in Saccharomyces cerevisiae. J Biosci Bioeng. 2016;121(6):665–671.
- Negoro H, Sakamoto M, Kotaka A, et al. Mutation in the peroxin-coding gene PEX22 contributing to high malate production in Saccharomyces cerevisiae. J Biosci Bioeng. 2018;125(2):211–217.
- Takahashi T, Ohara Y, Sawatari M, et al. Isolation and characterization of sake yeast mutants with enhanced isoamyl acetate productivity. J Biosci Bioeng. 2017;123(1):71–77.
- Takahashi T, Ohara Y, Sueno K. Breeding of a sake yeast mutant with enhanced ethyl caproate productivity in sake brewing using rice milled at a high polishing ratio. J Biosci Bioeng. 2017;23(6):707–713.
- Aikawa M, Suizu T, Ichikawa E, et al. Breeding of higher malic acid-productive mutants from Saccharomyces cerevisiae Kyokai No.7 (K7). Hakko-Kogaku. 1992;70(6):473–477. (in Japanese).
- Inokoshi J, Tomoda H, Hashimoto H, et al. Cerulenin-resistant mutants of Saccharomyces cerevisiae with an altered fatty acid synthase gene. Mol Gen Genet. 1994;244(1):90–96.
- Horie K, Oba T, Motomura S, et al. Breeding of a low pyruvate-producing sake yeast by isolation of a mutant resistant to ethyl alpha-transcyanocinnamate, an inhibitor of mitochondrial pyruvate transport. Biosci Biotechnol Biochem. 2010;74(4):843–847.
- Kadowaki M, Fujimaru Y, Taguchi S. Chromosomal aneuploidy improves the brewing characteristics of sake yeast. Appl Environ Microbiol. 2017;83(24):e01620–17.
- Tamura H, Okada H, Kume K, et al. Isolation of a spontaneous cerulenin-resistant sake yeast with both high ethyl caproate-producing ability and normal checkpoint integrity. Biosci Biotechnol Biochem. 2015;79(7):1191–1199.
- Goshima T, Nakamura R, Kume K, et al. Identification of a mutation causing a defective spindle-assembly checkpoint in high ethyl-caproate producing sake yeast strain K1801. Biosci Biotechnol Biochem. 2016;80(8):1657–1662.
- Ohnuki S, Okada H, Friedrich A, et al. Phenotypic diagnosis of lineage and differentiation during sake yeast breeding. G3 (Bethesda). 2017;7(8):2807–2820.
- Watanabe D, Kajihara T, Sugimoto Y, et al. Nutrient signalling via the TORC1-greatwall-PP2AB55δ pathway responsible for the high initial rates of alcoholic fermentation in sake yeast strains of Saccharomyces cerevisiae. Appl Environ Microbiol. 2019;85(1):e02083–18.
- Pavelka N, Rancati G, Zhu J, et al. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321–325.
- Mulla W, Zhu J, Li R. Yeast: a simple model system to study complex phenomena of aneuploidy. FEMS Microbiol Rev. 2014;38(2):201–212.
- Peter J, De Chiara M, Friedrich A, et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature. 2018;556(7701):339–344.
- Shimoi H, Hamazumi Y, Kawamura N, et al. Meiotic chromosomal recombination defect in sake yeasts. J Biosci Bioeng. 2018. DOI:10.1016/j.jbiosc.2018.07.027.
- Nakazawa N, Ashikari T, Goto N, et al. Partial restoration of sporulation defect in sake yeasts, Kyokai No. 7 and No. 9, by increased dosage of the IME1 gene. J Ferment Bioeng. 1992;73(4):265–270.
- Mori K, Kadooka C, Masuda C, et al. Genome sequence of Saccharomyces cerevisiae strain Kagoshima No. 2, used for brewing the Japanese distilled spirit shōchū. Genome Announc. 2017;5(41):pii:e01126-17.
- Kajiwara Y, Mori K, Tashiro K, et al. Genomic sequence of Saccharomyces cerevisiae BAW-6, a yeast strain optimal for brewing barley shochu. Genome Announc. 2018;6(14):pii: e00228-18.
- Futagami T, Kadooka C, Ando Y, et al. Multi-gene phylogenetic analysis reveals that shochu-fermenting Saccharomyces cerevisiae strains form a distinct sub-clade of the Japanese sake cluster. Yeast. 2017;34(10):407–415.