
ABSTRACT
Secondary metabolites produced by actinobacteria have diverse structures and important biological activities, making them a useful source of drug development. Diversity of the secondary metabolites indicates that the actinobacteria exploit various chemical reactions to construct a structural diversity. Thus, studying the biosynthetic machinery of these metabolites should result in discovery of various enzymes catalyzing interesting and useful reactions. This review summarizes our recent studies on the biosynthesis of secondary metabolites from actinobacteria, including the biosynthesis of nonproteinogenic amino acids used as building blocks of nonribosomal peptides, the type II polyketide synthase catalyzing polyene scaffold, the nitrous acid biosynthetic pathway involved in secondary metabolite biosynthesis and unique cytochrome P450 catalyzing nitrene transfer. These findings expand the knowledge of secondary metabolite biosynthesis machinery and provide useful tools for future bioengineering.
Graphical abstract
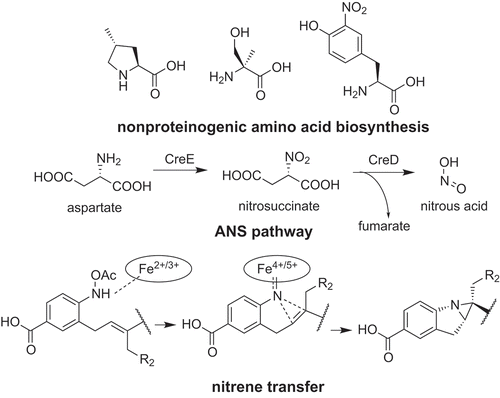
Biosynthesis of secondary metabolites produced by actinobacteria exploits various chemical reactions to construct a structural diversity.
Actinobacteria are high-GC content Gram-positive bacteria producing secondary metabolites with a wide variety of structures, including polyketides, terpenes, ribosomal and nonribosomal peptides. Since Waksman discovered streptomycin, an antituberculosis drug, form one of the actinobacteria, Streptomyces griseus, actinobacteria have been considered an important source of drug [Citation1]. In addition to streptomycin, highly important drugs such as an antibiotic (vancomycin), an immunosuppressant (FK-506) and an anticancer drug (doxorubicin) were also discovered from actinobacteria. The important feature of this class of microorganisms is its ability to synthesize diverse structures of secondary metabolites and the chemistry proceeding inside the cells. This interesting feature has attracted many scientists to study their biosynthetic machinery of secondary metabolites. Advances in biotechnology and the accumulation of knowledge in biosynthetic machinery have enabled researchers to develop a way to construct “unnatural” natural products by engineering biosynthetic pathways in microorganisms [Citation2–Citation5]. They also opened a way to discover novel natural products and enzymes based on genome databases [Citation1,Citation6,Citation7]. These technologies are important for drug development and construction of a sustainable society. However, many compounds with an unclear biosynthetic logic remain. This review highlights our recent study on the biosynthesis of secondary metabolites produced by actinobacteria.
Biosynthesis of nonribosomal peptides
Nonribosomal peptides (NRPs) are synthesized not from ribosome but by the enzymes called nonribosomal peptide synthetases (NRPSs) [Citation8]. This class of natural products includes important antibiotics such as vancomycin, daptomycin, and β-lactam. The important feature of NRPs is their ability to use nonproteinogenic amino acids as building blocks. By using such building blocks, NRPSs are able to produce peptides with diverse structures and bioactivities.
Griselimycin (1) is the cyclic NRP produced by Streptomyces. It possesses anti-tuberculosis activity by targeting DnaN containing DNA polymerase ()) [Citation9]. It uses two or three (2S,4R)-methylprolines (2) as building blocks ()) [Citation10]. Genome sequencing revealed that the griselimycin biosynthetic gene cluster (gri cluster) consists of three NRPS-encoding genes and four genes (griE, F, G, H) encoding enzymes predicted to be responsible for methylproline biosynthesis. Through heterologous expression and in vitro analysis of the recombinant enzymes, we have revealed the biosynthetic pathway of (2S,4R)-methylproline (2) ()). First, the α-ketoglutarate dependent dioxygenase, GriE, catalyzes stereospecific hydroxylation of leucine to synthesize (2S,4R)-5-hydroxyleucine (3). Then, GriF catalyzes NAD+ dependent dehydrogenation of (2S,4R)-5-hydroxyleucine (3) to synthesize (2S,4R)-4-methylglutamate-5-semialdehyde (4). This compound undergoes spontaneous cyclization and is converted to (3R,5S)-3-methyl-Δ1-pyrroline-5-carboxylic acid (5). GriH catalyzes the reduction of this compound to synthesize (2S,4R)-methylproline (2). This reaction seemed to also be catalyzed by ProC from proline biosynthesis because the heterologous expression of griEFGH showed that GriH is not essential for (2S,4R)-methylproline (2) synthesis in vivo. The most important reaction in the biosynthetic pathway is leucine hydroxylation, which was catalyzed by GriE, because this enzyme determines the stereochemistry of methyl group.
Figure 1. Biosynthesis of nonproteinogenic amino acids used as building blocks of nonribosomal peptides. Biosynthesis pathways of methylproline (2) (a), α-methyl-l-serine (8) (b) and nitrotyrosine (10) (c) used as building blocks of griselimycin (1), JBIR-34 (6), −35 (7) and rufomycin (9), respectively, are shown. KG, alpha-ketoglutarate; 5,10-MTHF, 5,10-methylenetetrahydrofolate; THF, tetrahydrofolate; SC, succinate.
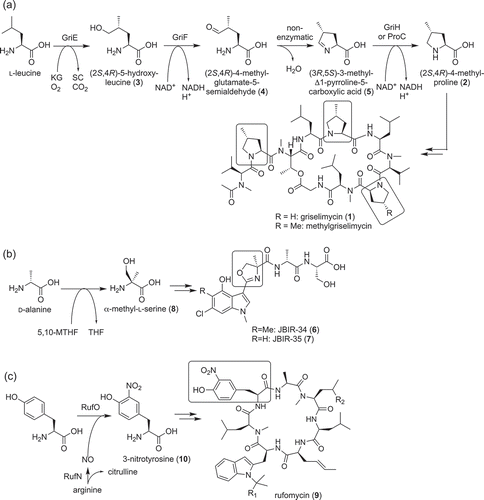
JBIR-34 (6), and −35 (7) are linear peptides isolated from Streptomyces sp. Sp080513GE-23 ()) [Citation11]. These compounds possess weak radical scavenging activity. Interestingly, these compounds consist of a 4-methyloxazoline ring. In general, an oxazoline ring with a methyl group in a peptide has a methyl group at position five because it is synthesized from threonine. Thus, this ring was predicted to be biosynthesized from nonproteinogenic amino acid. The in vivo feeding study indicated that this ring is derived from α-methyl-l-serine (8) synthesized by hydroxymethylation of d-alanine ()) [Citation12]. The biosynthetic gene cluster of JBIR-34 (6), and −35 (7) (fmo cluster) harbored one gene encoding putative glycine/serine hydroxymethyltransferase homolog, FmoH. The substrate specificity of recombinant FmoH was analyzed in vitro by using an inverse reaction. This experiment showed that FmoH prefers α-methyl-l-serine (8) as its substrate and it synthesized d-alanine from α-methyl-l-serine (8) using tetrahydrofolate as a co-factor. FmoH also catalyzed the conversion of l-serine to glycine, but this activity was much lower. These results indicated that FmoH catalyzes the biosynthesis of α-methyl-l-serine (8) from d-alanine in vivo ()). The substrate specificities of NRPSs in the fmo cluster (FmoA1, A2, A3, A4, A5) was also analyzed in vitro. This experiment showed that one of these NRPSs, FmoA3, prefers α-methyl-l-serine (8) as its substrate. According to these results, the biosynthetic mechanism of 4-methyloxazoline ring was proposed [Citation12].
Rufomycin (9) [Citation13], which is also known as ilamycin [Citation14–Citation16], is a cyclic peptide that also possesses anti-mycobacterial activity produced by Streptomyces atratus ATCC 14046. As nonproteinogenic building blocks, biosynthesis of rufomycin (9) seemed to require N-dimethylallyltryptophan, trans-2-crotylglycine, and 3-nitrotyrosine (10). By genome sequencing Streptomyces atratus ATCC 14046, the rufomycin biosynthetic gene cluster (ruf cluster) was discovered [Citation13]. This cluster encoded one large NRPS that consists of seven modules (RufT), one polyketide synthase (PKSs) and several cytochrome P450s. Interestingly, one of the cytochrome P450 (RufO) encoding genes is located adjacent to nitric oxide synthase (NOS, RufN) gene. Because direct nitration of the aromatic group was reported to be catalyzed by TxtD (NOS) and TxtE (cytochrome P450) encoded next to each other [Citation17], RufO and RufN were predicted to have similar functions with them and be responsible for nitration of tyrosine. Mutation of rufO genes abolished the ability to produce rufomycins. In addition, supplementation of 3-nitrotyrosine (10) to this mutant restored the production of rufomycins (9). Further, in vitro analysis clearly showed that RufO catalyzes nitration of tyrosine by using nitric oxide. RufO is the second cytochrome P450 reported to catalyze direct nitration of an aromatic group.
Biosynthetic machinery discovered from polyketide biosynthesis
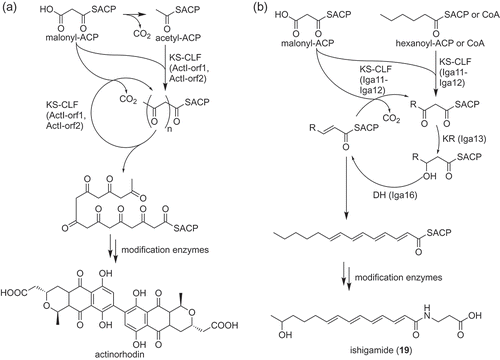
Polyketides are natural products synthesized by condensation of acetyl units by an enzyme called polyketide synthase. Because polyketides produced by actinobacteria also have important activities represented by erythromycin and doxorubicin, polyketides are also an important group of natural products to be studied. The structural diversity of polyketides usually arises from the structural diversity of substrates used for initiation and elongation, the number of elongation reactions and post PKS modification steps [Citation18].
Streptazone E (11) is the polyketide with the cyclopenta[b]pyridine scaffold isolated from Streptomyces sp. MSC090213JE08 ()) [Citation19]. This compound was also isolated from Streptomyces sp. NEAU-Z4 [Citation20]. The biosynthetic gene cluster of streptazone E (stz) was discovered by draft genome sequencing [Citation19]. This cluster contained three genes encoding PKSs. Interestingly, one PKS harbored C-terminal reductive releasing domain, which catalyzes the release of the polyketide chain as an aldehyde [Citation21]. This indicated that the amino group of streptazone E (11) is incorporated via aldehyde formation and reductive amination of an aldehyde. The biosynthetic pathway of strepazone E (11) was further analyzed by the mutagenesis of putative biosynthetic genes. As a result, the biosynthetic pathway of streptazone E (11) was predicted as in ). The most interesting reaction in this pathway is its five-membered ring formation coupled with an epoxide ring opening, which was predicted to be catalyzed by StzE and StzF, putative cyclases belonging to the nuclear transport factor 2-like superfamily ()). To date, the mechanism of this cyclization reaction is not clear because the in vitro analysis of StzE and F have not been established.
Figure 2. Biosynthesis of polyketides, streptazone E (11) (a), JBIR-76 (12), and −77 (13) (b) and fogacins (15–17) (c).
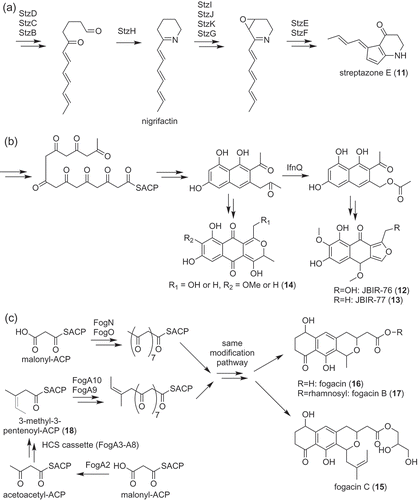
JBIR-76 (12) and −77 (13) are aromatic polyketides with an isofuranonaphthoquinone structure [Citation22]. Most of the multicyclic aromatic polyketides produced by actinobacteria are synthesized by type II PKSs ()) [Citation23]. Therefore, JBIR-76 (12) and −77 (13) were also expected to be biosynthesized by type II PKSs. The genome sequencing of the producer strain, Streptomyces sp. RI-77, showed that this strain has two type II PKS gene clusters. One was highly similar to the UT‐X26 biosynthetic gene cluster [Citation24]. Therefore, the other cluster was expected to be responsible for JBIR-76 (12) and −77 (13) biosynthesis. According to the structure of JBIR-76 (12) and −77 (13), a C–C bond cleavage was expected to be involved in their biosynthesis. IfnQ, which was a putative flavin-dependent Baeyer-Villiger monooxygenase, was the candidate enzyme for catalyzing the reaction. Inactivation of the ifnQ gene in the producer abolished the production of JBIR-76 (12) and −77 (13). Instead, this strain accumulated putative shunt products with 6-6-6 tricyclic rings (14) ()). The core structures of these compounds consist of 15 carbon atoms indicating that IfnQ is responsible for the C–C bond cleavage of the polyketide intermediate (the core structures of JBIR-76 (12) and −77 (13) consist of 13 carbon atoms). In vitro analysis of IfnQ was carried out using bicyclic model substrates mimicking an octaketide intermediate. Among the used substrates, IfnQ only used 1-(2-naphthyl)acetone as a substrate and synthesized 2‐naphthalenemethyl acetate. Thus, the IfnQ‐catalyzed C–C bond cleavage reaction was shown to be the key step in the biosynthesis of isofuranonaphthoquinone scaffolds ()).
Recently, a novel aromatic polyketide with a unique structure was isolated from Actinoplanes missouriensis [Citation25]. This aromatic polyketide was named fogacin C (15) because it was a derivative of fogacin (16), which was isolated from Streptomyces strain Tü 6319 ()) [Citation26]. Fogacin (16) and fogacin B (O-rhamnosyl-fogacin, 17) were also isolated from A. missouriensis ()). Fogacin C (15) was unique because biosynthesis of this compound apparently required β-alkylation of the polyketide chain ()). β-alkylation of a polyketide chain is relatively rare compared with α-alkylation [Citation27,Citation28]. In addition, β-alkylation had been reported only from the type I PKS system but not from type II PKS system. Therefore, the biosynthetic machinery of fogacin C (15) was expected to be interesting. By analyzing the genome sequence of A. missouriensis [Citation29], a biosynthetic gene cluster encoding two sets of ketosynthase (KS, FogN, and Fog A10) and chain length factor (CLF, FogO, and FogA9), was discovered. KS and CLF are core components of the type II PKS system and their heterodimers are responsible for elongation of the polyketide chain. In addition, this gene cluster possessed genes called hydroxymethylglutaryl-CoA synthase (HCS) cassette, which is responsible for β-alkylation of the polyketide chain [Citation27]. The function of the two sets of KS–CLF and the HCS cassette was analyzed by a gene inactivation experiment and heterologous expression. As a result, the HCS cassette with some genes was shown to be responsible for synthesizing the starter substrate, 3-methyl-3-pentenoyl thioester (18), via β-alkylation of the acetoacetyl unit ()). In addition, two sets of KS–CLF were shown to have a different starter substrate specificity; FogA10–FogA9 prefers a 3-methyl-3-pentenoyl unit (18) while FogN–FogO prefers an acetyl unit, a typical starter substrate for the type II PKS system ()). Both polyketides synthesized by these KS–CLF heterodimers seemed to be modified by the same modification pathway and converted to corresponding fogacin derivatives. This phenomenon indicated that A. missouriensis developed a combinatorial biosynthesis-like strategy to enhance the structural diversity of aromatic polyketides.
For a long time, Type II PKS system had been believed to be only responsible for the biosynthesis of aromatic polyketides ()) [Citation23]. However, the type II PKS system, which catalyzes polyene biosynthesis, was discovered and characterized in vitro from the ishigamide biosynthesis pathway ()) [Citation30,Citation31]. Ishigamide (19) is a polyketide with polyene structure discovered by genome mining that exploited forced expression of the Streptomyces antibiotic regulatory protein (SARP) family transcriptional regulator [Citation30]. Through the gene inactivation experiment, this compound was shown to be biosynthesized by a cluster containing type II PKS (iga cluster). Phylogenetic analysis showed that the KS (Iga11) and the CLF (Iga12) encoded in the cluster were clearly different from KSs and CLFs responsible for aromatic polyketide biosynthesis [Citation30]. To prove that this type II PKS is actually responsible for biosynthesis of the polyene structure, the subunits of the PKSs (Iga11–Iga12 heterodimer; ketoreductase, Iga13; dehydratase, Iga16; acyl carrier protein, Iga10) were prepared using E. coli and analyzed in vitro. As a result, polyene synthesis was successfully reconstituted in vitro [Citation31]. This experiment was the first proof of the existence of “highly reducing” type II PKS catalyzing polyene formation.
Figure 3. Biosynthesis of an aromatic polyketide and a polyene polyketide by type II PKS systems. Overview of biosynthetic pathways of actinorhodin (a) and ishigamide (19) (b) are shown as examples, respectively.
Discovery of a nitrous acid biosynthetic pathway specific for secondary metabolism in actinobacteria
Some actinobacteria produce natural products containing a diazo group [Citation32,Citation33]. Because of the high activity of the diazo group, most of these compounds have interesting activities. However, the biosynthesis of the diazo group remained unclear. Recent studies showed that biosynthesis of the diazo group requires a unique nitrous acid biosynthesis pathway. This pathway was first discovered from the biosynthetic pathway of cremeomycin (20) produced by Streptomyces cremeus () [Citation34,Citation35]. The biosynthetic gene cluster of cremeomycin (cre cluster) was identified by cosmid library construction, colony hybridization based on the genes responsible for 3-amino-4-hydroxybenzoic acid biosynthesis [Citation36], and heterologous expression. Gene inactivation in the heterologous expression strain showed that two enzymes, CreE (flavin-dependent monooxygenase) and CreD (lyase) are responsible for diazo group formation. In vitro analysis of the recombinant proteins showed that CreE catalyzes the oxidation of aspartate to synthesize nitrosuccinate (21) and CreD catalyzes nitrous acid (22) formation coupled with fumarate formation from nitrosuccinate (21). The nitrous acid (22) produced by these two enzymes is used for diazo group synthesis [Citation35]. Although the enzymes responsible for the diazo group are not clear to date, Waldman et al. provided preliminary evidence that the AMP-dependent ligase, CreM, catalyzes this reaction [Citation37]. Interestingly, an operon containing creE and creD homologs is widely distributed among actinobacteria, including Kitasatospora, Nocardia and Mycobacterium. Furthermore, many of these genes seem to be surrounded by genes presumably related to secondary metabolism, indicating that this nitrous acid biosynthetic pathway is involved in the biosynthesis of other secondary metabolites. Indeed, by analyzing the creE and creD homologs from Streptomyces davawensis, novel desferrioxamine derivatives, with unusual five-membered rings, was discovered (desferrioxamine I, 23–25, ) [Citation38]. This pathway is now called the ANS (aspartate-nitrosuccinate) pathway and is proposed to be a secondary metabolism specific nitrous acid (22) biosynthesis pathway. Genes encoding the ANS pathway were also discovered from biosynthetic gene clusters of fosfazinomycin (26) [Citation39,Citation40], kinamycin (27) [Citation40,Citation41], calcimycin [Citation42] and triacsin (28) [Citation43], and proved to be involved in the biosynthesis of these compounds. Those reports cited in the previous sentence indicated that the ANS pathway is involved in the biosynthesis of various secondary metabolites. Further study on the ANS pathway should provide important insights into role of nitrous acid (22) in secondary metabolism.
Figure 4. Secondary metabolite specific nitrous acid biosynthetic pathway discovered from actinobacteria and natural products synthesized by this pathway.
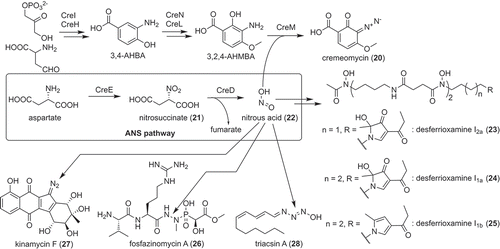
Not only the function of the ANS pathway in secondary metabolism but also the enzymes involved in this pathway are intriguing. CreD catalyzed nitrous acid (22) formation only when CreE was catalyzing oxidation in the same reaction mixture in vitro. This indicated that there might be some interaction between these enzymes [Citation35]. CreE is also a unique flavin-dependent monooxygenase that catalyzes the tandem three-step oxidation of an amino group. CreD is the first enzyme belonging to the aspartase/fumarase superfamily reported to catalyze the elimination of nitrous acid (22). Although most mechanisms of these features remained unclear, the reaction mechanism of nitrous acid (22) elimination catalyzed by CreD was elucidated by X-ray crystallography and site-directed mutagenesis [Citation44]. Interestingly, CreD was proposed to use Arg325 as a catalytic acid to stimulate the elimination of nitrous acid (22). In general, arginine is not preferred as a catalytic acid because of its high pKa value. CreD seemed to lower the pKa value of the arginine side chain with a cationic environment provided by His237, which is located behind Arg325 and is presumably stabilized as a cation by its interaction with Asp109 [Citation44].
Biosynthesis of other metabolites
Benzastatin derivatives are bacterial alkaloids composed of p-aminobenzoic acid (PABA, 29) moiety and geranyl moiety ()) [Citation45,Citation46]. Benzastatin derivatives can be further divided into linear (30, 31, 32), indoline (33, 34) and tetrahydroquinoline (35) according to their structures. Although their structural properties and biological activities are interesting, the biosynthesis of benzastatin derivatives had been unclear. The biosynthetic gene cluster of benzastatin derivatives was first analyzed by heterologous expression and gene inactivation using a heterologous host [Citation47]. As a result, two enzymes, BezA (methyltransferase) and BezC (cytochrome P450), were revealed to be responsible for the modification of geranyl moiety. Furthermore, three enzymes, BezJ (N-oxygenase), BezG (acetyltransferase) and BezE (cytochrome P450), were required for cyclization [Citation47].
Figure 5. Biosynthesis of benzastatin derivatives (a) and the key reaction involved in its bicyclic ring formation (b).
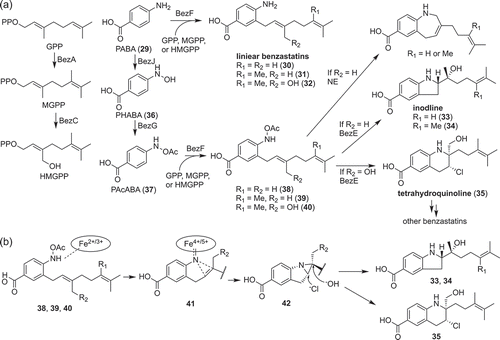
In vitro analysis of BezA and BezC showed that these enzymes catalyze sequential methylation and hydroxylation of geranyl pyrophosphate, respectively ()) [Citation47]. The in vitro analysis of the cyclization reaction was rather difficult because BezJ activity could not be detected in vitro despite extensive efforts. Therefore, the function of BezJ was predicted to be the N-hydroxylation of PABA by a feeding experiment; production of cyclized benzastatins was restored by feeding N-hydroxy-p-aminobenzoic acid (PHABA, 36) to the ΔbezJ strain. Then, BezG was proved to catalyze the O-acetylation of PHABA to synthesize N-acetyoxy-p-aminobenzoic acid (PAcABA, 37). According to these results, BezE was predicted to catalyze cyclization using geranylated PAcABA as a substrate (38–40) ()). To prove this hypothesis, biosynthesis of cyclized benzastatins was reconstituted in vitro and BezE was successfully shown to catalyze the cyclization reaction to synthesize both the indoline (33, 34) and tetrahydroquinoline (35) scaffolds ()) [Citation47]. This reaction presumably proceeds via formation of iron nitrenoid (41) stabilized by coordination to the heme iron, nitrene transfer to the adjacent double bond, which results in an aziridine ring (42), and an aziridine ring opening by a nucleophilic attack of HO− or Cl− ()) [Citation47]. This nonoxidative reaction catalyzed by BezE is completely different from the typical reaction catalyzed by cytochrome P450, which usually catalyzes monooxygenation. Although there is no cytochrome P450 that catalyzes nitrene transfer from the natural world, Arnold and co-workers developed artificial cytochrome P450s, which can catalyze nitrene transfer [Citation48]. BezE probably catalyzes cyclization in a manner similar to these artificial enzymes.
Various actinobacteria produce prenyl indole derivatives [Citation49–Citation52]. Through physicochemical screening, A. missouriensis was also discovered to produce a prenyl indole derivative, 3-hydroxy-6-dimethylallylindolin-2-one [Citation51]. A. missouriensis also produced 6-dimethylallyltryptophan and 6-dimethylallylindole. By searching the complete genome of A. missouriensis [Citation29], homolog (iptA−Am) of indole prenyltransferases (IptA and SCO7467) [Citation52,Citation53] encoded next to a putative tryptophanase gene (tnaA−Am) was discovered. Through an in vivo gene inactivation experiment and in vitro analysis of recombinant enzymes, IptA−Am and TnaA−Am were shown to catalyze the dimethylallylation of tryptophan and the conversion of 6-dimethylallyltryptophan to 6-dimethylallylindole, respectively. Although the oxidases that convert 6-dimethylallylindole to 3-hydroxy-6-dimethylallylindolin-2-one remained undiscovered, 6-dimethylallylindole was proved to be the intermediate of 3-hydroxy-6-dimethylallylindolin-2-one during a feeding experiment.
Future perspective
In the last decades, stimulated by the advance in next-generation sequencer and genetic engineering technology, the knowledge of biosynthesis of natural products has dramatically increased. However, the knowledge available to date seems to be “the tip of the iceberg”. In this review, novel biosynthetic machineries, which were discovered recently by our group, were described. These include the discovery and identification of the type II PKS that catalyze polyene formation [Citation30,Citation31], a nitrous acid biosynthetic pathway specific for secondary metabolism [Citation35,Citation38,Citation44] and the cytochrome P450 that catalyzes the nitrene transfer reaction [Citation47]. These systems seemed to be distributed among actinobacteria but have been overlooked for a long time. It is expected that novel enzymatic chemistry can still be discovered by studying the biosynthesis of secondary metabolites produced by actinobacteria. Some of the enzymes described in this review might be useful for industrial purposes. RufO is a useful enzyme that can add a nitro group directly to the aromatic ring [Citation13] and the nitrene transfer reaction is useful for synthesizing reactive species. The high activity of a diazo group enables it to be a useful reagent for 1,3-dipolar cycloaddition, nucleophilic substitution, and carbene formation in synthetic organic chemistry [Citation35,Citation37]. Detailed analysis of the enzymes and engineering may provide a novel tool for producing useful compounds including “unnatural” natural products. Our studies significantly expanded the knowledge of the biosynthesis of secondary metabolites and provided important information for future bioengineering for combinatorial biosynthesis.
Acknowledgments
These studies were carried out at The University of Tokyo, and Saarladn University. I am very grateful to Professors Sueharu Horinouchi, Yasuo Ohnishi, and Rolf Müller for their support and helpful discussions. I thank all my co-workers for their cooperation. I also would like to thank all the members of the Laboratory of Fermentation who participated in this work. This research was supported by Japan Society for the Promotion of Science [grant number 25850048, 25108706, 17H05432 and 18H02144], the NC-CARP project from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), CREST, JST, JSPS A3 Foresight Program and Amano Enzyme Inc.
Disclosure statement
No potential conflict of interest was reported by the author.
Additional information
Funding
References
- Katz L, Baltz RH. Natural product discovery: past, present, and future. J Ind Microbiol Biotechnol. 2016;43:155–176.
- Katsuyama Y, Hirose Y, Funa N, et al. Precursor-directed biosynthesis of curcumin analogs in Escherichia coli. Biosci Biotechnol Biochem. 2010;74:641–645.
- Horinouchi S. Mining and polishing of the treasure trove in the bacterial genus Streptomyces. Biosci Biotechnol Biochem. 2007;71:283–299.
- Katsuyama Y, Ohnishi Y, Horinouchi S. Production of dehydrogingerdione derivatives in Escherichia coli by exploiting a curcuminoid synthase from Oryza sativa and a β-oxidation pathway from Saccharomyces cerevisiae. Chembiochem Eur J Chem Biol. 2010;11:2034–2041.
- Katsuyama Y, Funa N, Miyahisa I, et al. Synthesis of unnatural flavonoids and stilbenes by exploiting the plant biosynthetic pathway in Escherichia coli. Chem Biol. 2007;14:613–621.
- Choi -S-S, Katsuyama Y, Bai L, et al. Genome engineering for microbial natural product discovery. Curr Opin Microbiol. 2018;45:53–60.
- Ziemert N, Alanjary M, Weber T. The evolution of genome mining in microbes - a review. Nat Prod Rep. 2016;33:988–1005.
- Süssmuth RD, Mainz A. Nonribosomal peptide synthesis-principles and prospects. Angew Chem Int Ed Engl. 2017;56:3770–3821.
- Kling A, Lukat P, Almeida DV, et al. Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science. 2015;348:1106–1112.
- Lukat P, Katsuyama Y, Wenzel S, et al. Biosynthesis of methyl-proline containing griselimycins, natural products with anti-tuberculosis activity. Chem Sci. 2017;8:7521–7527.
- Motohashi K, Takagi M, Shin-Ya K. Tetrapeptides possessing a unique skeleton, JBIR-34 and JBIR-35, isolated from a sponge-derived actinomycete, Streptomyces sp. Sp080513GE-23. J Nat Prod. 2010;73:226–228.
- Muliandi A, Katsuyama Y, Sone K, et al. Biosynthesis of the 4-methyloxazoline-containing nonribosomal peptides, JBIR-34 and −35, in Streptomyces sp. Sp080513GE-23. Chem Biol. 2014;21:923–934.
- Tomita H, Katsuyama Y, Minami H, et al. Identification and characterization of a bacterial cytochrome P450 monooxygenase catalyzing the 3-nitration of tyrosine in rufomycin biosynthesis. J Biol Chem. 2017;292:15859–15869.
- Takita T, Naganawa H, Maeda K, et al. The structures of ilamycin and ilamycin B2. J Antibiot. 1964;17:129–131.
- Takita T, Naganawa H, Maeda K, et al. The structural difference among ilamycin, ilamycin cl and ilamycin C2. J Antibiot. 1965;18:135–136.
- Ma J, Huang H, Xie Y, et al. Biosynthesis of ilamycins featuring unusual building blocks and engineered production of enhanced anti-tuberculosis agents. Nat Commun. 2017;8:391.
- Barry SM, Kers JA, Johnson EG, et al. Cytochrome P450–catalyzed L-tryptophan nitration in thaxtomin phytotoxin biosynthesis. Nat Chem Biol. 2012;8:814–816.
- Miyanaga A. Structure and function of polyketide biosynthetic enzymes: various strategies for production of structurally diverse polyketides. Biosci Biotechnol Biochem. 2017;81:2227–2236.
- Ohno S, Katsuyama Y, Tajima Y, et al. Identification and characterization of the Streptazone E biosynthetic gene cluster in Streptomyces sp. MSC090213JE08. Chembiochem Eur J Chem Biol. 2015;16:2385–2391.
- Liu Q-F, Wang J-D, Wang X-J, et al. Two new piperidine alkaloids from Streptomyces sp. NEAU-Z4. J Asian Nat Prod Res. 2013;15:221–224.
- Mullowney MW, McClure RA, Robey MT, et al. Natural products from thioester reductase containing biosynthetic pathways. Nat Prod Rep. 2018;35:847–878.
- Katsuyama Y, Sone K, Satou R, et al. Involvement of the Baeyer-Villiger monooxygenase IfnQ in the biosynthesis of isofuranonaphthoquinone scaffold of JBIR-76 and −77. Chembiochem Eur J Chem Biol. 2016;17:1021–1028.
- Hertweck C, Luzhetskyy A, Rebets Y, et al. Type II polyketide synthases: gaining a deeper insight into enzymatic teamwork. Nat Prod Rep. 2007;24:162–190.
- Feng Z, Kallifidas D, Brady SF. Functional analysis of environmental DNA-derived type II polyketide synthases reveals structurally diverse secondary metabolites. Proc Natl Acad Sci U S A. 2011;108:12629–12634.
- Sato K, Katsuyama Y, Yokota K, et al. Involvement of β-alkylation machinery and two sets of ketosynthase-chain length factors in the biosynthesis of fogacin polyketides in Actinoplanes missouriensis. Chembiochem. 2019;20:1039–1050.
- Radzom M, Zeeck A, Antal N, et al. Fogacin, a novel cyclic octaketide produced by Streptomyces strain Tü 6319. J Antibiot. 2006;59:315–317.
- Calderone CT. Isoprenoid-like alkylations in polyketide biosynthesis. Nat Prod Rep. 2008;25:845–853.
- Bretschneider T, Heim JB, Heine D, et al. Vinylogous chain branching catalysed by a dedicated polyketide synthase module. Nature. 2013;502:124–128.
- Yamamura H, Ohnishi Y, Ishikawa J, et al. Complete genome sequence of the motile actinomycete Actinoplanes missouriensis 431(T) (= NBRC 102363(T)). Stand Genomic Sci. 2012;7:294–303.
- Du D, Katsuyama Y, Onaka H, et al. Production of a novel amide-containing polyene by activating a cryptic biosynthetic gene cluster in Streptomyces sp. MSC090213JE08. Chembiochem Eur J Chem Biol. 2016;17:1464–1471.
- Du D, Katsuyama Y, Shin-Ya K, et al. Reconstitution of a Type II polyketide synthase that catalyzes polyene formation. Angew Chem Int Ed Engl. 2018;57:1954–1957.
- Nawrat CC, Moody CJ. Natural products containing a diazo group. Nat Prod Rep. 2011;28:1426–1444.
- Le Goff G, Ouazzani J. Natural hydrazine-containing compounds: biosynthesis, isolation, biological activities and synthesis. Bioorg Med Chem. 2014;22:6529–6544.
- McGuire JN, Wilson SR, Rinehart KL. Cremeomycin, a novel cytotoxic antibiotic from Streptomyces cremeus. Structure elucidation and biological activity. J Antibiot. 1995;48:516–519.
- Sugai Y, Katsuyama Y, Ohnishi Y. A nitrous acid biosynthetic pathway for diazo group formation in bacteria. Nat Chem Biol. 2016;12:73–75.
- Suzuki H, Ohnishi Y, Furusho Y, et al. Novel benzene ring biosynthesis from C(3) and C(4) primary metabolites by two enzymes. J Biol Chem. 2006;281:36944–36951.
- Waldman AJ, Balskus EP. Discovery of a diazo-forming enzyme in cremeomycin biosynthesis. J Org Chem. 2018;83:7539–7546.
- Hagihara R, Katsuyama Y, Sugai Y, et al. Novel desferrioxamine derivatives synthesized using the secondary metabolism-specific nitrous acid biosynthetic pathway in Streptomyces davawensis. J Antibiot. 2018;71:911–919.
- Huang Z, Wang -K-KA, van der Donk WA. New insights into the biosynthesis of fosfazinomycin. Chem Sci. 2016;7:5219–5223.
- Wang -K-KA, Ng TL, Wang P, et al. Glutamic acid is a carrier for hydrazine during the biosyntheses of fosfazinomycin and kinamycin. Nat Commun. 2018;9:3687.
- Liu X, Liu D, Xu M, et al. Reconstitution of kinamycin biosynthesis within the heterologous host Streptomyces albus J1074. J Nat Prod. 2018;81:72–77.
- Wu H, Liang J, Wang J, et al. Cezomycin is activated by CalC to its ester form for further biosynthesis steps in the production of calcimycin in Streptomyces chartreusis NRRL 3882. Appl Environ Microbiol. 2018;84:e00586-18.
- Twigg FF, Cai W, Huang W, et al. Identifying the biosynthetic gene cluster for triacsins with an N-hydroxytriazene moiety. Chembiochem Eur J Chem Biol. 2019;20:1–6.
- Katsuyama Y, Sato Y, Sugai Y, et al. Crystal structure of the nitrosuccinate lyase CreD in complex with fumarate provides insights into the catalytic mechanism for nitrous acid elimination. FEBS J. 2018;285:1540–1555.
- Kim WG, Kim JP, Kim CJ, et al. Benzastatins A, B, C, and D: new free radical scavengers from Streptomyces nitrosporeus 30643. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J Antibiot. 1996;49:20–25.
- Motohashi K, Nagai A, Takagi M, et al. Two novel benzastatin derivatives, JBIR-67 and JBIR-73, isolated from Streptomyces sp. RI18. J Antibiot. 2011;64:281–283.
- Tsutsumi H, Katsuyama Y, Izumikawa M, et al. Unprecedented cyclization catalyzed by a cytochrome P450 in benzastatin biosynthesis. J Am Chem Soc. 2018;140:6631–6639.
- Brandenberg OF, Fasan R, Arnold FH. Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr Opin Biotechnol. 2017;47:102–111.
- Motohashi K, Irie K, Toda T, et al. Studies on terpenoids produced by actinomycetes. 5-dimethylallylindole-3-carboxylic acid and A80915G-8"-acid produced by marine-derived Streptomyces sp. MS239. J Antibiot. 2008;61:75–80.
- Sasaki T, Igarashi Y, Ogawa M, et al. Identification of 6-prenylindole as an antifungal metabolite of Streptomyces sp. TP-A0595 and synthesis and bioactivity of 6-substituted indoles. J Antibiot. 2002;55:1009–1012.
- Satou R, Izumikawa M, Katsuyama Y, et al. Isolation, structural elucidation and biosynthesis of 3-hydroxy-6-dimethylallylindolin-2-one, a novel prenylated indole derivative from Actinoplanes missouriensis. J Antibiot. 2014;67:231–236.
- Takahashi S, Takagi H, Toyoda A, et al. Biochemical characterization of a novel indole prenyltransferase from Streptomyces sp. SN-593. J Bacteriol. 2010;192:2839–2851.
- Ozaki T, Nishiyama M, Kuzuyama T. Novel tryptophan metabolism by a potential gene cluster that is widely distributed among actinomycetes. J Biol Chem. 2013;288:9946–9956.