
ABSTRACT
A concise, unified total synthesis of the two prenylated aromatic polyketides amorfrutins A and C, which exhibit various medicinally important biological profiles such as antimicrobial, PPARγ modulating and quorum sensing inhibitory activities, has been achieved from commercially available 3,5-dimethoxybenzaldehyde in 38% and 10% overall yields through nine and ten steps, respectively. The key transformation for the synthesis of amorfrutin A was the Claisen rearrangement of a mono-O-(1,1-dimethylallyl)resorcinol derivative to install the C3-prenyl substituent, while that for the synthesis of amorfrutin C was the double Claisen rearrangement of a di-O-(1,1-dimethylallyl)resorcinol derivative to introduce the two prenyl groups at the C3 and C5 positions all at once.
Graphical abstract
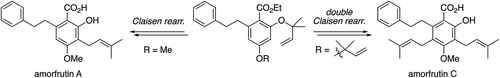
Unified total synthesis of amorfrutins A and C via the Claisen rearrangement.
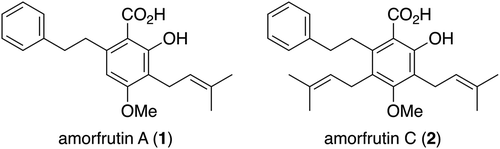
Amorfturin A (1) () is a polyketide–terpenoid hybrid meroterpene isolated first from the bastard indigobush Amorpha fruitcosa [Citation1] and later from the two perennial herbs Glycyrrhiza acanthocarpa [Citation2] and G. foetida [Citation3]. It belongs to the amorfrutin family of natural products [Citation4] and structurally features a salicylic acid nucleus substituted at the C3, C4, and C6 positions with a prenyl, a methoxy, and a 2-phenylethyl group, respectively. The fully substituted aromatic compound amorfrutin C (2), a congener of amorfrutin A (1) with an additional prenyl substituent at the C5 position, is also a member of this family recently discovered from G. foetida [Citation5]. Until now, a variety of biological activities have been reported for 1, 2, and other members of this family, including antimicrobial [Citation1,Citation6], NF-κB (nuclear transcription factor-κB) inhibitory [Citation7], PPARγ (peroxisome proliferator-activated receptor gamma) modulating [Citation3,Citation8–Citation10], anti-HCV (hepatitis C virus) [Citation11], and quorum sensing inhibitory properties [Citation12]. These medicinally important biological activities of amorfrutins coupled with their unique poly-substituted aromatic architectures have prompted considerable synthetic efforts toward 1 and 2, which culminated in six total syntheses of amorfrutin A (1) [Citation8,Citation11–Citation16] as well as two total syntheses of amorfrutin C (2) [Citation17,Citation18].
Figure 1. Structures of amorfrutins A (1) and C (2).
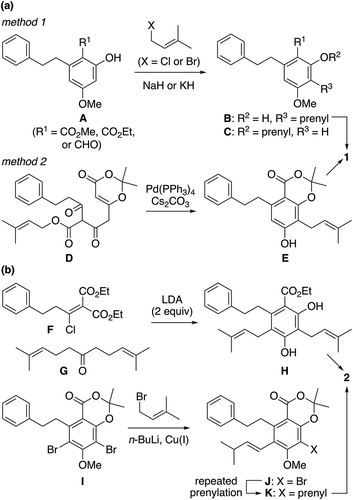
The previously reported total syntheses of 1 can be categorized into 2 types according to the method to install the C3-prenyl substituent ()): C-prenylation of resorcinol derivative A (R1 = CO2Me, CO2Et, or CHO) (method 1) and Pd(0)-catalyzed migratory prenylation–aromatization of substituted dioxinone D (method 2). The C-prenylation of A gave desired C-prenylation product B in moderate yields of 38–58% together with substantial amounts of O-prenylated byproduct C [Citation8,Citation11,Citation12,Citation14]. The indirect conversion of A (R = CO2Me) into B via montmorillonite K10-catalyzed rearrangement of C, which could be prepared O-selectively from A in 92% yield, also resulted in a modest yield of 38% over two steps [Citation16]. The unique and efficient decarboxylative prenyl migration–aromatization approach (method 2) successfully converted D into desired product E in 62% isolated yield [Citation13]. As for amorfrutin C (2), the Sauer group employed base-induced condensation between F and G to obtain 56% yield of H with two prenyl groups properly and efficiently installed ()) [Citation17]. In Xie’s synthesis of 2, the two prenyl groups were introduced into m-dibromobenzene derivative I in a stepwise manner, but the yield of the two-step sequence leading to K via J was 36%, leaving room for improvement [Citation18].
Scheme 1. Previous methods to install the C3-prenyl substituent of 1 (a) and C3- and C5-prenyl groups of 2 (b).
The preceding studies were, as described above, directed toward the synthesis of each of amorfrutins A and C, and no strategy enabling the synthesis of the two natural products in a unified manner has been reported so far. In this article, we describe a unified total synthesis of the two amorfrutins using the Claisen rearrangement as the key reaction.
Results and discussion
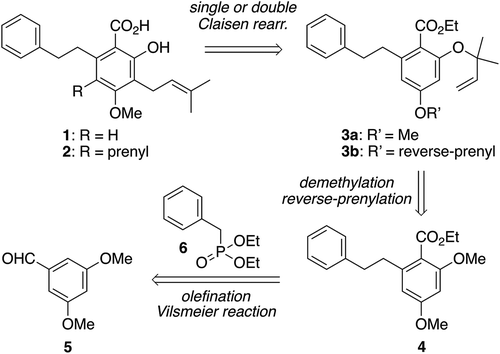
Our retrosynthetic analysis of amorfrutins A (1) and C (2) is outlined in . The target molecule 1 would be obtainable via the Claisen rearrangement of aryl ether 3a bearing a 1,1-dimethylallyl (reverse-prenyl) group [Citation19] on the oxygen atom at the C2 position, while the 3,5-diprenylated congener 2 would be derived through the double Claisen rearrangement of 2,4-di-O-(reverse-prenyl) ether 3b. The aryl ethers, 3a and 3b, would be prepared from m-dimethoxybenzene derivative 4 as a common precursor by selective demethylation of the methyl ether moiety ortho to the ester group followed by reverse-prenylation and demethylation of the two methyl ether moieties at the C2 and C4 positions followed by double reverse-prenylation, respectively. The dimethoxy benzoate 4 should be prepared from known aromatic aldehyde 5 via its olefination with phosphonate 6 followed by hydrogenation to install the 2-phenylethyl side chain and the Vilsmeier-Haack reaction to introduce a carbonyl functionality at the C1 position.
Scheme 2. Retrosynthetic analysis of amorfrutins A (1) and C (2).
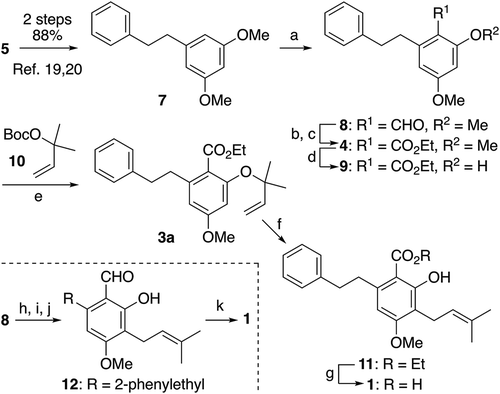
We first undertook the synthesis of amorfrutin A (1) (). Known bibenzyl compound 7, obtained by the Horner–Wadsworth–Emmons olefination of 5 with 6 and subsequent catalytic hydrogenation of the resulting stilbene intermediate according to the literature [Citation20,Citation21], was subjected to the Vilsmeier–Haack reaction to give 8 as a single formylation product in 81% yield [Citation22]. The Pinnick oxidation of the aromatic aldehyde 8 followed by esterification of the carboxylic acid thus produced afforded 4 in an excellent yield of 93% for the 2 steps. Selective mono-demethylation of the methyl ether moiety at the C2 position of 4 with BBr3 (1.1 equiv) provided phenol 9 in 77% yield [Citation23]. Treatment of 9 with carbonate 10 in the presence of a catalytic amount of Pd(PPh3)4 and 4Å molecular sieves in THF effected a Tsuji–Trost-type etherification [Citation24], furnishing the aryl reverse-prenyl ether 3a almost quantitatively. It is worth mentioning that the addition of pulverized activated 4Å molecular sieves was necessary to obtain reproducibly high yields of 3a. The Claisen rearrangement of 3a to 11 was performed in 77% yield by heating a solution of 3a in xylene under microwave irradiation conditions. Finally, saponification of the ester 11 furnished amorfrutin A (1) in 94% yield. It should be noted that we first attempted the oxidation of o-hydroxy aldehyde 12 prepared from 8 in 3 steps, but all attempted conditions including the Jones and the Pinnick oxidation conditions, 1-Me-AZADO/PhI(OAc)2, and AgNO3/KOH resulted in low yields of up to 32%.
Scheme 3. Synthesis of amorfrutin A (1).
Reagents and conditions: (a) POCl3, DMF, –4ºC to rt, 21 h, 81%; (b) NaClO2, NaH2PO4, 2-methyl-2-butene, THF/H2O (6:1), rt, 12 h; (c) K2CO3, EtI, MeCN, 46ºC, 40 min, 93% from 8; (d) BBr3, CH2Cl2, –78ºC, 4 h, 77%; (e) Pd(PPh3)4, 4Å MS, THF, –20ºC, 19.5 h, 98%; (f) microwave irradiation, o-xylene, reflux, 70 min, 77%; (g) KOH, EtOH/H2O (1:1), 80ºC, 6 h, 94%; (h) BBr3, CH2Cl2, –78ºC to rt, 2.5 h, 90%; (i) 10, Pd(PPh3)4, 4Å MS, THF, 0°C, 16.5 h, quant; (j) o-xylene, reflux, 2 h, 77%; (k) NaClO2, NaH2PO4, 2-methyl-2-butene, THF/H2O (12:1), rt, 6 h, 32%.
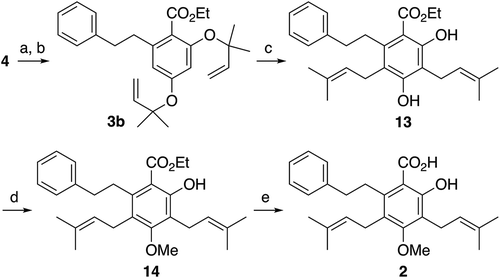
The synthesis of amorfrutin C (2) was implemented by a five-step sequence from the common intermediate 4 as shown in . Demethylation of both the C2 and C4 methoxy groups of 4 by treatment with an excess amount of BBr3 afforded a resorcinol intermediate in 75% yield, although it required a higher temperature (0°C) and a longer reaction time (36 h) as compared to those employed for mono-demethylation of 4 to afford 9 (–78°C, 4 h; see ). Reverse-prenylation of the two phenolic hydroxyls of the intermediate proceeded uneventfully to furnish a quantitative yield of 3b, which was then heated under microwave irradiation conditions to perform its double Claisen rearrangement to 13. This reaction gave a mixture of several products after complete consumption of the starting material 3b, from which the desired product 13 was successfully isolated in a moderate yield of 52%. Finally, selective O-methylation of the C4-OH group of 13 with MeI and K2CO3 in acetone [Citation17] followed by saponification of the resulting ester 14 completed the total synthesis of amorfrutin C (2). The spectroscopic data of the synthetic amorfrutin C (2) were in good agreement with those of natural product [Citation5,Citation18].
Scheme 4. Synthesis of amorfrutin C (2).
Reagents and conditions: (a) BBr3, CH2Cl2, 0ºC, 36 h, 75%; (b) Pd(PPh3)4, 10, 4Å MS, THF, –20ºC, 18 h, quant; (c) microwave irradiation, o-xylene, reflux, 3 h, 52%; (d) MeI, K2CO3, acetone, rt, 19 h, 50%; (e) KOH, EtOH/H2O (5:1), 70ºC, 70 h, 75%.
Conclusion
The concise total synthesis of amorfrutin A (1) has been accomplished from commercially available 3,5-dimethoxybenzaldehyde 5 in 38% overall yield by a nine-step sequence (or from the known compound 7 in 48% overall yield through 7 steps) that involves the Claisen rearrangement of the mono-reverse-prenylated intermediate 3a as the key step. Amorfrutin C (2), 5-prenylated congener of 1, has also been synthesized in ten steps from 5 in 10% overall yield by exploiting the double Claisen rearrangement of 2,4-di-O-reverse-prenylated intermediate 3b, which was derived from them-dimethoxybenzene derivertive 4, a common synthetic precursor of the two amorfrutins. Synthetic efforts toward other members of this class of natural products are now in progress and will be reported in due course.
Experimental
General procedure
IR spectra were recorded by a Jasco FT/IR-4100 spectrometer using an ATR (ZnSe) attachment. NMR spectra were recorded with TMS as an internal standard in CDCl3 by a Varian 400-MRTT spectrometer (400 MHz for 1H and 100 MHz for 13C) or a Varian 600TT spectrometer (600 MHz for 1H and 150 MHz for 13C) unless otherwise stated. Optical rotation values were measured with a Jasco P-2200 polarimeter. Mass spectra were obtained with Jeol JMS-700 spectrometer operated in the FAB mode. Merck silica gel 60 (63–200 μm) was used for column chromatography unless otherwise stated. Analytical thin-layer chromatography was performed using Merck silica gel 60 F254 plates (0.25 mm thick). Solvents for reactions were distilled prior to use: THF from Na and benzophenone; CH2Cl2 and DMF from CaH2. All air- or moisture-sensitive reactions were conducted under a nitrogen atmosphere unless otherwise stated.
2,4-dimethoxy-6-(2-phenylethyl)benzaldehyde (8)
A stock solution of the Vilsmeier reagent (ca. 2.0 m in DMF) was prepared in advance from DMF (31.5 mL) and POCl3 (7.20 mL, 77.3 mmol). The solution (18 mL, 36 mmol) was added dropwise to a stirred solution of 1,3-dimethoxy-5-(2-phenylethyl) benzene (3.53 g, 14.6 mmol) in DMF (24.0 mL) at – 4°C. The reaction mixture was warmed to room temperature and stirred for 21 h. The reaction mixture was quenched with satd aq NH4Cl while cooling with an ice bath and extracted three times with EtOAc. The extract was successively washed with 5% aq LiCl and brine, dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by SiO2 column chromatography (hexane/EtOAc = 9:1) to give aldehyde 8 (3.17 g, 81%) as a white solid, whose spectral data were identical to those reported in the literature [Citation22].
Ethyl 2,4-dimethoxy-6-(2-phenylethyl)benzoate (4)
To a stirred solution of 8 (202 mg, 0.747 mmol) in THF/H2O (6:1, 7 mL) was successively added NaH2PO4 (258 mg, 2.15 mmol), 2-methyl-2-butene (800 μL, 7.55 mmol) and NaClO2 (75% dispersion in H2O, 270 mg, 2.24 mmol) at room temperature. After stirring for 12 h, the reaction mixture was diluted with brine and extracted three times with CH2Cl2. The extract was dried (Na2SO4), filtered and concentrated in vacuo and the residue was taken up in MeCN (2.20 mL). To the solution was successively added K2CO3 (310 mg, 2.24 mmol) and EtI (300 µL, 3.75 mmol) at room temperature. The mixture was warmed to 46ºC and stirred for 40 min. The mixture was diluted with brine at room temperature and extracted three times with EtOAc. The extract was dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by SiO2 column chromatography (hexane/EtOAc = 4:1) to give 4 (219 mg, 93% from 8) as a colorless oil. IR: υmax 1723 (s), 1602 (s), 1587 (m), 1496 (m); 1H NMR (400 MHz, CDCl3): δ 1.37 (3H, t, J = 7.1 Hz), 2.83–2.93 (4H, m), 3.75 (3H, s), 3.81 (3H, s), 4.38 (2H, q, J = 7.2 Hz), 6.24 (1H, d, J = 2.2 Hz), 6.33 (1H, d, J = 2.2 Hz), 7.16–7.22 (3H, m), 7.26–7.31 (2H, m); 13C NMR (100 MHz, CDCl3): δ 14.3, 36.1, 37.6, 55.3, 55.9, 61.1, 96.6, 105.9, 116.6, 126.0, 128.36 (2C), 128.42 (2C), 141.5, 141.7, 158.0, 161.2, 168.3; HRMS (FAB): m/z calcd for C19H23O4, 315.1591; found, 315.1599 ([M + H]+).
Ethyl 2-hydroxy-4-methoxy-6-(2-phenylethyl)-benzoate (9)
To a stirred solution of 4 (128 mg, 0.407 mmol) in CH2Cl2 (4.10 mL) was added dropwise a solution of BBr3 (1.0 m in CH2Cl2, 440 µL, 0.44 mmol) at – 78ºC. After stirring for 4 h at the same temperature, the reaction mixture was quenched with satd aq NaHCO3 and extracted three times with CH2Cl2. The extract was washed with brine, dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by SiO2 column chromatography (hexane/EtOAc = 10:1) to give 9 (94.0 mg, 77%) as a white solid. Mp 51–52 ºC; IR: υmax 1651 (vs), 1616 (s), 1322 (m), 1257 (s); 1H NMR (400 MHz, CDCl3): δ 1.39 (3H, t, J = 7.1 Hz), 2.84–2.90 (2H, m), 3.18–3.24 (2H, m), 3.77 (3H, s), 4.44 (2H, q, J = 7.2 Hz), 6.25 (1H, d, J = 2.7 Hz), 6.36 (1H, d, J = 2.7 Hz), 7.15–7.23 (3H, m), 7.27–7.32 (2H, m), 11.9 (1H, s, OH); 13C NMR (100 MHz, CDCl3): δ 14.3, 38.0, 38.4, 55.3, 61.4, 99.2, 104.8, 110.9, 126.0, 128.2 (2C), 128.3 (2C), 141.8, 146.7, 163.9, 165.6, 171.4; HRMS (FAB): m/z calcd for C18H21O4, 301.1434; found, 301.1438 ([M + H]+).
Ethyl 4-methoxy-2-[(2-methylbut-3-en-2-yl)oxy]-6-(2-phenylethyl)benzoate (3a)
To a stirred solution of 9 (83.0 mg, 0.276 mmol) and tert-butyl (2-methylbut-3-en-2-yl) carbonate (10) (180 mg, 0.966 mmol) in degassed THF (550 µL) was added pulverized 4Å molecular sieves (276 mg) at room temperature. After cooling to – 20ºC, Pd(PPh3)4 (16.0 mg, 0.0138 mmol) was added portionwise and the resulting suspension was stirred for 19.5 h. The reaction mixture was filtered through a pad of Celite and the filtrate was concentrated in vacuo. The residue was purified by SiO2 column chromatography (pentane/Et2O = 8:1) to give 3a (100 mg, 98%) as a colorless oil. IR: υmax 3025 (m), 1725 (s), 1602 (s), 1579 (m), 1199 (m), 1158 (s); 1H NMR (400 MHz, CDCl3): δ 1.37 (3H, t, J = 7.1 Hz), 1.46 (6H, s), 2.80–2.91 (4H, m), 3.68 (3H, s), 4.35 (2H, q, J = 7.2 Hz), 5.16 (1H, dd, J = 0.9, 10.9 Hz), 5.23 (1H, dd, J = 0.9, 17.6 Hz), 6.19 (1H, dd, J = 10.9, 17.7 Hz), 6.29 (1H, d, J = 2.3 Hz), 6.58 (1H, d, J = 2.3 Hz), 7.15–7.21 (3H, m), 7.24–7.30 (2H, m); 13C NMR (100 MHz, CDCl3): δ 14.4, 26.9 (2C), 36.1, 37.6, 55.2, 60.9, 80.6, 103.7, 107.6, 113.4, 120.6, 125.9, 128.3 (2C), 128.4 (2C), 140.9, 141.6, 144.8, 154.9, 160.1, 168.7; HRMS (FAB): m/z calcd for C23H29O4, 369.2060; found, 369.2067 ([M + H]+).
Ethyl 2-hydroxy-4-methoxy-3-(3-methylbut-2-en-1-yl)-6-(2-phenylethyl)benzoate (11)
A solution of 3a (99.0 mg, 0.269 mmol) in o-xylene (2.00 mL) in a microwave reaction vessel was heated to reflux under irradiation of microwave (300 W, CEM Discover Microwave System) for 70 min. The reaction mixture was concentrated in vacuo and the residue was purified by SiO2 column chromatography (hexane/EtOAc = 20:1) to give 11 (76 mg, 77%) as a white solid. Mp 64ºC; IR: υmax 3027 (m), 1727 (s), 1650 (w), 1602 (s), 1459 (m), 1158 (s); 1H NMR (400 MHz, CDCl3): δ 1.39 (3H, t, J = 7.2 Hz), 1.66–1.69 (3H, m), 1.78 (3H, br s), 2.85–2.91 (2H, m), 3.19–3.25 (2H, m), 3.33 (2H, d, J = 7.0 Hz), 3.77 (3H, s), 4.44 (2H, q, J = 7.1 Hz), 5.17–5.23 (1H, m), 6.16 (1H, s), 7.15–7.23 (3H, m), 7.24–7.32 (2H, m), 11.8 (1H, s, OH); 13C NMR (100 MHz, CDCl3): δ 14.3, 17.8, 22.0, 25.8, 38.2, 38.8, 55.4, 61.3, 105.3, 106.0, 115.2, 122.4, 125.9, 128.31 (2C), 128.34 (2C), 131.6, 141.9, 143.9, 161.0, 161.9, 171.7; HRMS (FAB): m/z calcd for C23H28O4Na, 391.1880; found, 391.1882 ([M+ Na]+).
2-hydroxy-4-methoxy-3-(3-methylbut-2-en-1-yl)-6-(2-phenylethyl)benzoic acid: amorfrutin a (1)
To a stirred solution of 11 (53.0 mg, 0.144 mmol) in a mixed solvent of EtOH and H2O (1:1, 1.40 mL) was added KOH (343 mg, 6.11 mmol) at room temperature. The reaction mixture was warmed to 80ºC, stirred for 5 h, and then concentrated in vacuo. The residue was acidified with 2 m HCl (pH 3) and extracted five times with CHCl3. The extract was washed with brine, dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by SiO2 column chromatography (hexane/EtOAc = 10:1) to give 1 (46 mg, 94%) as a white solid. An analytical sample was recrystallized from hexane/EtOAc to give NMR spectroscopically pure 1 as colorless prisms. Mp 151–152ºC; IR: υmax 3025 (m), 2926 (br), 1627 (s), 1610 (s), 1455 (m), 1267 (s); 1H NMR (400 MHz, CDCl3): δ 1.68 (3H, d, J = 0.9 Hz), 1.79 (3H, br s), 2.88–2.97 (2H, m), 3.22–3.31 (2H, m), 3.35 (2H, d, J = 7.0 Hz), 3.79 (3H, s), 5.17–5.24 (1H, m), 6.22 (1H, s), 7.17–7.23 (3H, m), 7.27–7.33 (2H, m), 11.6 (1H, br s, OH);13C NMR (100 MHz, CDCl3): δ 17.8, 22.0, 25.8, 38.1, 39.2, 55.5, 103.6, 106.5, 115.3, 122.1, 125.9, 128.4 (2C), 128.5 (2C), 131.8, 141.9, 145.8, 162.1, 162.9, 176.0; HRMS (FAB): m/z calcd for C21H24O4Na, 363.1567; found, 363.1570 ([M+ Na]+).
Ethyl 2,4-bis[(2-methylbut-3-en-2-yl)oxy]-6-(2-phenylethyl)benzoate (3b)
To a stirred solution of 4 (3.05 g, 9.70 mmol) in CH2Cl2 (60.0 mL) was added dropwise a solution of BBr3 (1.0 m in CH2Cl2, 50.0 mL, 48.5 mmol) at 0 ºC. After stirring for 36 h, the reaction mixture was quenched with satd aq NaHCO3 and extracted four times with CHCl3. The extract was successively washed with water and brine, dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by SiO2 column chromatography (hexane/EtOAc = 8:1) to give ethyl 2,4-dihydroxy-6-(2-phenylethyl)benzoate (2.08 g, 75%) as a pale yellow solid. Mp 89–90ºC; IR: υmax 3381 (br), 1652 (s), 1618 (s), 1495 (m), 1314 (s), 1262 (s); 1H NMR (400 MHz, CDCl3): δ 1.39 (3H, t, J = 7.2 Hz), 2.84–2.90 (2H, m), 3.17–3.23 (2H, m), 4.44 (2H, q, J = 7.2 Hz), 5.10–5.13 (1H, m), 6.18 (1H, d, J = 2.6 Hz), 6.30 (1H, d, J = 2.6 Hz), 7.15–7.23 (3H, m), 7.25–7.33 (2H, m) 11.8 (1H, s, OH);13C NMR (100 MHz, CDCl3): δ 14.1, 37.8, 38.1, 61.6, 101.6, 104.9, 111.3, 125.9, 128.1 (2C), 128.3 (2C), 141.6, 147.6, 160.6, 164.9, 171.5; HRMS (FAB): m/z calcd for C17H19O4, 287.1278; found, 287.1288 ([M + H]+). To a stirred solution of the dihydroxy benzoate obtained above (517 mg, 1.81 mmol) and tert-butyl (2-methylbut-3-en-2-yl) carbonate (2.06 g, 11.1 mmol) in THF (3.60 mL) was added pulverized 4Å molecular sieves (1.38 g) at room temperature. After cooling to – 20ºC, Pd(PPh3)4 (209 mg, 0.181 mmol) was added portionwise and the resulting suspension was stirred for 18 h. The reaction mixture was filtered through a pad of Celite and the filtrate was concentrated in vacuo. The residue was purified by SiO2 column chromatography (pentane/Et2O = 9:1, containing 1% Et3N) to give 3b (770 mg, quant) as a pale yellow oil. IR: υmax 3025 (w), 1730 (s), 1602 (s), 1264 (m); 1H NMR (400 MHz, CDCl3): δ 1.34–1.39 (9H, m), 1.43 (6H, s), 2.76–2.89 (4H, m), 4.35 (2H, q, J = 7.2 Hz), 5.06–5.21 (4H, m), 6.03 (1H, dd, J = 10.7, 17.8 Hz), 6.13 (1H, dd, J = 10.9, 17.6 Hz), 6.41 (1H, d, J = 2.1 Hz), 6.65 (1H, d, J = 2.1 Hz), 7.13–7.19 (3H, m), 7.23–7.29 (2H, m);13C NMR (100 MHz, CDCl3): δ 14.3, 26.86 (2C), 26.90 (2C), 35.7, 37.4, 60.8, 79.6, 80.4, 109.5, 113.1, 113.2, 115.4, 121.4, 125.8, 128.27 (2C), 128.34 (2C), 139.9, 141.6, 144.0, 144.5, 154.0, 156.7, 168.8; HRMS (FAB): m/z calcd for C27H35O4, 423.2530; found, 423.2539 ([M + H]+).
Ethyl 2,4-dihydroxy-3,5-bis(3-methylbut-2-en-1-yl)-6-(2-phenylethyl)benzoate (13)
A stirred solution of 3b (808 mg, 1.91 mmol) in o-xylene (2.0 mL) in a microwave reaction vessel was heated to reflux under irradiation of microwave (300 W, CEM Discover Microwave System) for 3 h. The reaction mixture was concentrated in vacuo and the residue was purified two times by SiO2 column chromatography (hexane/EtOAc = 9:1 and then CH2Cl2/benzene = 1:1) to give 13 (420 mg, 52%) as a pale yellow oil. IR: υmax 3403 (br), 3020 (w), 1647 (s), 1605 (m), 1311(s), 1267(s), 1199(s); 1H NMR (400 MHz, CDCl3): δ 1.36 (3H, t, J = 7.1 Hz), 1.71 (3H, d, J = 1.2 Hz), 1.75 (6H, s), 1.83 (3H, br s), 2.81–2.88 (2H, m), 3.19–3.26 (2H, m), 3.36 (2H, br d, J = 6.4 Hz), 3.45 (2H, br d, J = 7.0 Hz), 4.44 (2H, q, J = 7.2 Hz), 5.03–5.09 (1H, m), 5.22–5.29 (1H, m), 6.02 (1H, s), 7.18–7.25 (3H, m), 7.28–7.34 (2H, m), 11.7 (1H, s, OH);13C NMR (100 MHz, CDCl3): δ 14.4, 17.9, 18.0, 22.4, 25.1, 25.7, 25.8, 32.6, 37.1, 61.5, 105.8, 112.3, 119.3, 121.7, 122.9, 125.9, 128.0 (2C), 128.4 (2C), 133.3, 134.9, 141.1, 142.1, 158.3, 160.0, 172.0; HRMS (FAB): m/z calcd for C27H34O4Na, 445.2349; found, 445.2355 ([M+ Na]+).
Ethyl 2-hydroxy-4-methoxy-3,5-bis(3-methylbut-2-en-1-yl)-6-(2-phenylethyl)benzoate (14)
To a stirred solution of 13 (82.0 mg, 0.194 mmol) in acetone (1.90 mL) was added portionwise K2CO3 (26.0 mg, 0.188 mmol) at room temperature. After stirring for 5 min, the reaction mixture was cooled to – 1ºC. To the mixture was added MeI (12.0 μL, 0.193 mmol) and the mixture was warmed to room temperature and stirred for 19 h. The reaction mixture was concentrated in vacuo and the residue was diluted with a mixed solvent of H2O, hexane and Et2O (1:1:1). The mixture was neutralized with 1 m HCl and extracted three times with Et2O. The extract was dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by SiO2 column chromatography (hexane/EtOAc = 24:1) to give 14 (42 mg, 50%) as a colorless oil. IR: υmax 3382 (br), 1651 (s), 1317 (s), 1264 (s), 1198 (s); 1H NMR (400 MHz, CDCl3): δ 1.36 (3H, t, J = 7.1 Hz), 1.66 (3H, d, J = 1.3 Hz), 1.70 (3H, d, J = 1.2 Hz), 1.71 (3H, d, J = 0.9 Hz), 1.79 (3H, d, J = 0.8 Hz), 2.78–2.85 (2H, m), 3.19–3.26 (2H, m), 3.34 (2H, d, J = 6.1 Hz), 3.39 (2H, d, J = 6.6 Hz), 3.71 (3H, s), 4.45 (2H, q, J = 7.2 Hz), 4.99–5.06 (1H, m), 5.22–5.28 (1H, m), 7.17–7.24 (3H, m), 7.27–7.33 (2H, m), 11.2 (1H, s, OH);13C NMR (100 MHz, CDCl3): δ 14.3, 17.9, 18.0, 23.5, 25.3, 25.6, 25.7, 32.4, 37.3, 61.5, 61.7, 109.7, 121.2, 122.8, 124.2, 125.9, 126.1, 128.0 (2C), 128.3 (2C), 131.5, 131.7, 141.4, 142.1, 160.1, 161.4, 171.6; HRMS (FAB): m/z calcd for C28H36O4Na, 459.2506; found, 459.2510 ([M+ Na]+).
2-hydroxy-4-methoxy-3,5-bis(3-methylbut-2-en-1-yl)-6-(2-phenylethyl)benzoic acid: odfamorfrutin C (2)
To a stirred solution of 14 (51.0 mg, 0.117 mmol) in a mixed solvent of EtOH and H2O (5:1, 1.2 mL) was added KOH (34.0 mg, 0.606 mmol) at room temperature. The mixture was warmed to 70ºC and stirred for 70 h. After cooling to room temperature, the reaction mixture was concentrated in vacuo and the residue was acidified with 1 m HCl (pH 3). The mixture was extracted four times with CHCl3 and the extract was dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by SiO2 column chromatography (hexane/EtOAc = 3:1) to give 2 (36 mg, 75%) as a white solid. An analytical sample was recrystallized from hexane/EtOAc to give NMR spectroscopically pure 2 as colorless needles. Mp 112–113ºC; IR: υmax 3025 (m), 2970 (br), 1634 (s), 1590 (m), 1452 (m), 1259 (s); 1H NMR (600 MHz, CD3OD): δ 1.67 (6H, br s), 1.72 (3H, s), 1.78 (3H, s), 2.75–2.81 (2H, m), 3.16–3.21 (2H, m), 3.33–3.36 (4H, m), 3.67 (3H, s), 4.99–5.04 (1H, m), 5.20–5.25 (1H, m), 7.13–7.21 (3H, m), 7.25 (2H, t, J = 7.4 Hz);13C NMR (150 MHz, CD3OD): δ 18.0, 18.2, 24.3, 25.8, 25.9, 26.2, 34.4, 38.7, 61.9, 110.8, 122.0, 124.3, 125.7, 126.87, 126.93, 129.27, 129.32, 131.9 (2C), 132.1 (2C), 143.5, 143.8, 161.9, 162.5, 175.0; HRMS (FAB): m/z calcd for C26H32O4Na, 431.2193; found, 431.2198 ([M+ Na]+).
Author contribution
Y.O. and S.K. designed the synthetic route. Y.O. and S.K. wrote the manuscript. T.F. conducted the synthetic experiments with the aid of Y.O.
Supplemental.pdf
Download PDF (1.3 MB)Acknowledgments
We thank Ms. Yuka Taguchi (Tohoku University) for NMR and MS measurements.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Mitsher LA, Park YH, Al-Shamma A, et al. Amorfrutin A and B, bibenzyl antimicrobial agents from Amorpha fruticosa. Phytochemistry. 1981;20:781–785.
- Ghisalberti EL, Jefferies PR, McAdam D. Isoprenylated resorcinol derivatives from Glycyrrhiza acanthocarpa. Phytochemistry. 1981;20:1959–1961.
- Sauer S. Amorfrutins: a promising class of natural products that are beneficial to health. Chembiochem. 2014;15:1231–1238.
- Hanuš LO, Meyer SM, Muñoz E, et al. Phytocannabionids: a unified critical inventory. Nat Prod Rep. 2016;33:1357–1392.
- Weidner C, Rousseau M, Micikas RJ, et al. Amorfrutin C induces apoptosis and inhibits proliferation in colon cancer cells through targeting mitochondria. J Nat Prod. 2016;79:2–12.
- Muharini R, Díaz A, Ebrahim W, et al. Antibacterial and cytotoxic phenolic metabolites from the fruits of Amorpha fruticosa. J Nat Prod. 2017;80:169–180.
- Dat NT, Lee J-H, Lee K, et al. Phenolic constituents of Amorpha fruticosa that inhibit NF-κB activation and related gene expression. J Nat Prod. 2008;71:1696–1700.
- Weidner C, de Groot JC, Prasad A, et al. Amorfrutins are potent antidiabetic dietary natural products. Proc Natl Acad Sci USA. 2012;109:7257–7262.
- Weidner C, Wowro SJ, Freiwald A, et al. Amorfrutin B is an efficient natural peroxisome proliferator-activated receptor gamma (PPARγ) agonist with potent glucose-lowering properties. Diabetologia. 2013;56:1802–1812.
- Fuhr L, Rousseau M, Plauth A, et al. Amorfrutins are natural PPARγ agonists with potent anti-inflammatory properties. J Nat Prod. 2015;78:1160–1164.
- Ji X-Y, Chen J-H, Zheng G-H, et al. Design and synthesis of cajanine analogues against hepatitis C virus through down-regulating host chondroitin sulphate N-acetylgalactosaminyltransferase 1. J Med Chem. 2016;59:10268–10284.
- Xu X-J, Zeng T, Huang Z-X, et al. Synthesis and biological evaluation of cajaninstillbene acid and amorfrutins A and B as inhibitors of the Pseudomonas aeruginosa quorum sensing system. J Nat Prod. 2018;81:2621–2629.
- Laclef S, Anderson K, White AJP, et al. Total synthesis of amorfrutin A via a palladium-catalyzed migratory prenylation-aromatization sequence. Tetrahedron Lett. 2012;53:225–227.
- Song YY, He HG, Li Y, et al. A facile total synthesis of amorfrutin A. Tetrahedron Lett. 2013;54:2658–2660.
- Aidhen IS, Mukkamala R, Weidner C, et al. A common building block for the syntheses of amorfrutin and cajaninstilbene acid libraries toward efficient binding with peroxisome proliferator-activated receptors. Org Lett. 2015;17:194–197.
- Grandhi GS, Selvakumar J, Dana S, et al. Directed C-H bond functionalization: a unified approach to formal syntheses of amorfrutin A, cajaninstilbene acid, hydrangenol, and macrophyllol. J Org Chem. 2018;83:12327–12333.
- Sauer S, Weidner C, Kliem M, et al. Amorfrutin analogs as ppargamma-modulators. US Patent 2014177593, 2014.
- Miao Q, Li Y, Xu J, et al. First total synthesis of amorfrutin C and pseudo-amorfrutin A. Eur J Org Chem. 2018;2018:1443–1448.
- Marsden SP, Depew KM, Danishefsky SJ. Stereoselective total syntheses of amauromine and 5-N-acetylardeemin. A concise route to the family of “reverse-prenylated” hexahydropyrroloindole alkaloids. J Am Chem Soc. 1994;116:11143–11144.
- Wadsworth WS Jr. Synthetic applications of phosphoryl-stabillized anions. Org React. ( New York) 1977;25:74–253.
- Moodie LWK, Trepos R, Cervin G, et al. Prevention of marin biofouling using the natural allelopathic compound batatasin-III and synthetic analogues. J Nat Prod. 2017;80:2001–2011.
- Kumbhar VB, Joseph AR, Natu AD, et al. An efficient synthesis of bibenzylic oxygen heterocycles. J Chem Res. 2007;2007:590–593.
- Elix JA, Evans JE, Nash III TH. New depsides from Dimelaena lichens. Aust J Chem. 1988;41:1789–1796.
- Chantarasriwong O, Cho WC, Batova A, et al. Evaluation of the pharmacophoric motif of the caged Garcinia xanthones. Org Biomol Chem. 2009;7:4886–4894.