
ABSTRACT
(−)-Rotundone, a sesquiterpenoid that has a characteristic woody and peppery odor, is a key aroma component of spicy foodstuffs, such as black pepper and Australian Shiraz wine. (−)-Rotundone shows the lowest level of odor threshold in natural compounds and remarkably improves the quality of various fruit flavors. To develop a method for the synthesis of (−)-rotundone, we focused on non-heme Fe2+-chelates, which are biomimetic catalysts of the active center of oxygenases and enzymatic supply and regeneration of those catalysts. That is, we constructed a unique combination system composed of the oxidative synthesis of (−)-rotundone using the non-heme Fe2+-chelate catalyst, Fe(II)-EDTA, and the enzymatic supply and regeneration of Fe2+-chelate by ferric-chelate reductase, YqjH, from Escherichia coli. In addition, we improved the yield of (−)-rotundone by the application of cyclodextrin and glucose dehydrogenase to this system, and thus established a platform for efficient (−)-rotundone production.
Graphical Abstract
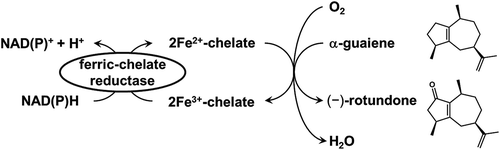
Catalytic synthesis of odor sesquiterpenoids by allylic oxidation using non-heme Fe2+-chelate catalyst and ferric-chelate reductase.
Sesquiterpenoids, which are oxygenated derivatives of sesquiterpenes, constitute the large diverse class among terpene compounds and exhibit various biological activities [Citation1–Citation3]. Among those sesquiterpenoids, we focused on (−)-rotundone [(3S,5R,8S)-5-isopropenyl-3,8-dimethyl-3,4,5,6,7,8-hexahydro-1(2H)-azulenone], which has a woody and peppery odor, with the lowest level of odor threshold in natural products (8 ng/L in water, 16 ng/L in red wine), and which is recognized as a key aroma component of spicy foodstuffs, such as black pepper and Australian Shiraz wine [Citation4,Citation5]. Furthermore, this compound has been identified in several fruits (e.g. grapefruit, orange, mango, and apple) and exhibits significant effects for the improvement of the overall qualities of flavors with a subthreshold addition [Citation6,Citation7]. However, the amount of (−)-rotundone found in natural sources is limited (from the ppb to ppm order); therefore, an effective synthesis method is required for its use in industrial applications [Citation4]. It is presumed that (−)-rotundone is oxidatively synthesized from the precursor sesquiterpene, α-guaiene, in Vitis vinifera (grapevine) [Citation8,Citation9]. And α-guaiene and its derivatives are widely distributed in natural sources, including guaiac wood oil or patchouli oil [Citation10,Citation11]. In terms of conventional chemical methods, allylic oxidation using chromate (VI) or manganite (VII) are regular candidates and advanced methods are under investigation [Citation12]. However, these methods require hazardous and non-ecological heavy metal catalysts and peroxidants, which should be replaced in order to carry out a more sustainable chemistry. Furthermore, there has been an increase in consumer demands for products of “naturalness” in the food industry, and the European regulation (EC No 1334/2008, article 3.2) defines natural flavoring substance that leads to the added value of natural products [Citation13–Citation15]. In these situations, biotechnological methods are a promising alternative. P450s are the most commonly used approach for the production of terpenoids, and several sesquiterpenoids, such as artemisinin and (+)-nootkatone, are already on the market [Citation2,Citation14,Citation16]. CYP71BE5 from V. vinifera has been reported to catalyze the oxidation of α-guaiene to (−)-rotundone [Citation8]. Additionally, the oxidation of α-guaiene with laccase [Citation17] or by aeration in an organic solvent and on cellulose filter paper [Citation18] has been reported. However, previous studies have experienced difficulties due to a low selectivity or productivity, and an effective method for the synthesis of (−)-rotundone has not yet been developed.
On the other hand, in the field of biomimetic catalysts, studies have been carried out on non-heme Fe2+-chelate catalysts, which mimic the active center of oxygenases, such as 2-oxoglutarate-dependent dioxygenase and pterin-dependent hydroxylase [Citation19,Citation20]. Iron-containing oxygenases have been found to generate high valent iron(IV)-oxo intermediates as active species using molecular oxygen and had catalyzed the oxygenation of substrates [Citation21]. Moreover, non-heme Fe2+-chelate catalysts are recognized to possess the same functions and the ability to catalyze various oxidative reactions containing aliphatic hydroxylation and alkene epoxidation [Citation22]. In particular, as the allylic oxidation of cyclohexene to cyclohexenol and cyclohexenone with non-heme Fe2+-chelate catalysts was reported [Citation23], we speculated that these methods could be applied to (−)-rotundone synthesis. Moreover, we designed a combination system composed of the catalytic synthesis of (−)-rotundone and the enzymatic supply and regeneration of Fe2+-chelate (). A vast number of microbes secrete iron-specific chelators, denoted as siderophores, which capture ferric ion and form Fe3+-chelates. Fe3+-chelates are then transported intracellularly and reduced enzymatically by ferric-chelate reductase to utilize ferrous iron [Citation24,Citation25]. Therefore, we focused on this ferric-chelate reductase for the supply and regeneration of Fe2+-chelate catalyst. Among ferric-chelate reductases, we selected YqjH from Escherichia coli since it has been well studied and is supposed to possess sufficient catalytic activity for this concept [Citation26,Citation27]. This system is comprised of the mutual reactions of metal-catalyzed oxygenation and the enzymatic supply and regeneration of catalyst, which provides a new insight into the synthesis of sesquiterpenoids.
Figure 1. Catalytic synthesis of odor sesquiterpenoid, (−)-rotundone, using non-heme Fe2+-chelate catalyst and ferric-chelate reductase.
Materials and methods
Reagents
NADH, NADPH, NAD+, and NADP+ were purchased from Nakarai Tesque (Kyoto, Japan). Catalase from bovine liver, HP-β-CD, glucose dehydrogenase from Bacillus sp., and formaldehyde test Wako were purchased from FUJIFILM Wako Pure Chemical (Osaka, Japan). Galvinoxyl free radical was purchased from Tokyo Chemical Industry (Tokyo, Japan). α-Guaiene, (−)-rotundone, and rotundol were synthesized and provided by T. Hasegawa (Kanagawa, Japan). All other chemicals were of analytical grade.
Screening assay
The screening for (−)-rotundone synthesis was performed with reaction mixtures (500 µL) containing 20 mM of each metal salt (e.g. FeSO4, FeCl3, Fe2(SO4)3, FeCl3, CuCl, CuI, CuSO4, ZnSO4, MnSO4, MgSO4, and CoSO4), 1% (vol/vol) Tween-80, 5 mM α-guaiene, and 50 mM KH2PO4-K2HPO4 buffer (pH 7.5). CuCl and CuI were saturated due to their low solubility. The reactions were carried out at 30°C for 1 h in glass vials and extracted using the double amount of ethyl acetate with 30 µg of (R)-carvone as an internal standard. The products were analyzed qualitatively with GC-MS and quantitated with HPLC.
Preparation of ferric-chelate reductase, YqjH
Purified YqjH was prepared in a similar manner as described previously [Citation27]. The gene yqjH was amplified by PCR using E. coli BL21(DE3) genomic DNA as a template and primers containing Nde I or Hind III restriction sites, respectively. The amplified fragments were digested and inserted into a pET-21a(+) vector (Novagen). E. coli BL21(DE3) cells were transformed with this plasmid. The recombinant cells were grown in 200 mL of LB medium containing 100 µg/ml ampicillin at 37°C until reaching an OD600 value of 0.7 ~ 1.0. Then, 1 mM IPTG was added and the cultivation was continued for 15 h at 25°C. The cells were harvested by centrifugation and resuspended in 50 mM HEPES-KOH buffer (pH 7.0). The cells were disrupted by sonication and centrifuged to obtain a supernatant. His-tagged YqjH was purified using a HisTrap HP 5 mL column equipped with ÄKTA Prime Plus and desalted using a PD-10 column, then eluted in 50 or 200 mM HEPES-KOH buffer (pH 7.0). The expression and purification of YqjH was confirmed by SDS-PAGE analysis.
Construction of ferric-chelate reductase system
(−)-Rotundone synthesis using ferric-chelate reductase, YqjH, was performed with a reaction mixture (500 µL) containing 5 mM α-guaiene, 1% Tween-80, 5 mM NADH or NADPH, 1 mM of each ferric iron (e.g. FeCl3, Fe(III)-dicitrate, or Fe(III)-EDTA) and 0.3 mg/mL purified YqjH in 50 mM HEPES-KOH buffer (pH 7.0). Fe(III)-dicitrate was prepared by mixing FeCl3 and trisodium citrate solution at a molar ratio of 1:2. Fe(III)-EDTA was prepared by dissolving EDTA-Na-Fe(III)∙3H2O. To verify the prerequisites for (−)-rotundone synthesis, cofactors (NADH, NADPH, and ferric iron) were subtracted from the reaction mixture one by one. The reactions were carried out at 30°C for 2 h, then stopped and extracted by adding the double amount of ethyl acetate with an internal standard. The consumption of NAD(P)H was monitored with a wavelength of 340 nm. The products were analyzed with GC-MS and HPLC.
Validation and improvement of ferric-chelate reductase system
To verify the reaction mechanism, the reactions (500 µL) were performed with 5 mM α-guaiene, 1% Tween-80, 5 mM NADPH, 1 mM Fe(III)-EDTA, 0.3 mg/mL purified YqjH in 50 mM HEPES-KOH buffer (pH 7.0), and 100 U/mL catalase or 1 mM galvinoxyl free radical. The reactions were carried out at 30°C for 2 h, then stopped and extracted by adding the double amount of ethyl acetate with an internal standard. To improve the efficiency of this system, the reactions (500 µL) were performed with 5 mM α-guaiene, 1% Tween-80, 5 mM NADH or NADPH, 1 mM Fe(III)-EDTA, 0.3 mg/mL purified YqjH in 200 mM HEPES-KOH buffer (pH 7.0), and 1% (weight/vol) hydroxypropyl-β-cyclodextrin (HP-β-CD). Then, the NAD(P)H was substituted with 1 mM NAD+ or NADP+, 100 mM glucose, and 0.01–1 U/mL glucose dehydrogenase (GlcDH). The reactions were carried out at 30°C for 2 h, then stopped and extracted by adding the double amount of ethyl acetate with an internal standard. The products were analyzed with GC-MS and HPLC. For the isolation and characterization of the by-product, preparative-scale reactions (20 ml) were performed with 5 mM α-guaiene, 1% Tween-80, 1 mM Fe(III)-EDTA, 1% HP-β-CD, 1 mM NAD+, 100 mM glucose, 0.1 U/ml GlcDH, and recombinant cells expressing YqjH (OD600 = 20, prepared as described above) in 200 HEPES-KOH buffer. The reactions were carried out at 30°C and extracted by adding the double amount of ethyl acetate with an internal standard. The reactions were conducted for several batches to accumulate the by-product for NMR analysis. To trace by-products in another means, C1 compounds (e.g. methanol, formaldehyde, and formic acid) were analyzed with GC-MS and formaldehyde was quantitated using a formaldehyde test Wako.
Product analysis
HPLC analysis was performed using a Prominence LC-20 system (Shimadzu, Kyoto, Japan) with a Wakosil-II 5C18 HG column (4.6 mm × 150 mm, FUJIFILM Wako Pure Chemical, Osaka, Japan). The extracted samples (10 µL) were analyzed with following separation program: gradient elution with acetonitrile 50–100% against ultrapure water for 10 min, column washing with acetonitrile 100% for 3 min, re-equilibration with acetonitrile 50% for 5 min at flow rate 1 mL/min. α-Guaiene and rotundol were detected at a wavelength of 220 nm, and (−)-rotundone and demethylated analog of (−)-rotundone were detected at a wavelength of 240 nm.
The GC-MS analysis was performed using a 7890 series GC system, 5977B Network Mass Selective Detector, with an HP-5ms Ultra Inert (30 m × 0.25 mm × 0.25 µm; Agilent Technologies, Santa Clara, CA, USA). The extracted samples (1 µL, split ratio 10:1, injection temp. 250°C) were analyzed using the following separation program: column oven initial temperature of 40°C for 1 min, 15°C/min gradient to 300°C at a flow rate of 1 mL/min He carrier gas.
1H and 13C NMR analysis was performed using a JNM-ECX400 (JEOL Resonance, Tokyo, Japan). (4S,7R)-7-isopropenyl-4-methyl-3,4,5,6,7,8-hexahydro-1(2H)-azulenone (demethylated analog of (−)-rotundone); 1H NMR (400 MHz, C6D6, Figure S7): δ = 0.81 (3H, d, J= 7.2 Hz), 1.27 (1H, m), 1.38 (1H, ddd, J= 4.0, 4.0, 9.6 Hz), 1.45 (1H, m), 1.64 (3H, s), 1.63–1.69 (2H, m), 1.82 (1H, m), 1.98 (1H, m), 2.03–2.07 (3H, m), 2.11 (1H, m), 2.97 (1H, d, J= 14.8 Hz), 4.75 (2H, d, J= 15.6 Hz). 13C NMR (100 MHz, C6D6, Figure S8): δ = 16.78, 20.70, 28.40, 29.87, 31.21, 32.42, 34.18, 36.72, 45.48, 109.20, 139.24, 150.68, 177.44, 207.23.
Results
Screening of the catalytic activity for (−)-rotundone synthesis
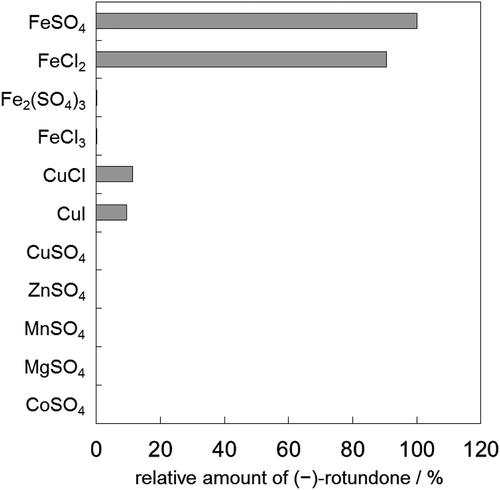
At the outset of this research, we performed a screening of microorganisms able to catalyze the allylic oxidation of α-guaiene to (−)-rotundone. However, contrary to our expectations, multiple strains including E. coli BL21(DE3) exhibited slight oxidizing activities against α-guaiene (data not shown). Therefore, we deduced that some ubiquitous components of microorganisms could affect oxidation. As such, we verified the effects of intracellular factors, such as metal ions and coenzymes. As a result of shifting targets, (−)-rotundone synthesis was confirmed using reduced form metal ions, in particular, Fe2+ ( and Figure S1). Ferrous iron is susceptible to molecular oxygen and is able to donate electrons and generate reactive oxygen species. Oxygenases with a Fe2+-chelate active center or biomimetic Fe2+-chelate catalysts utilize these properties for oxygenation [Citation21,Citation22], such that the formation of (−)-rotundone could be possible via these mechanisms. Since E. coli BL21(DE3) exhibited oxidizing activity, we focused on the ferric-chelate reductase, YqjH, as a candidate for supplying the Fe2+-chelate catalyst in (−)-rotundone synthesis.
Figure 2. Synthesis of (−)-rotundone using intracellular metal ions. Reaction mixtures contained α-guaiene and 20 mM of each metal salt. The amount of synthesized (−)-rotundone using FeSO4 was defined as 100%.
Construction of the catalytic system with non-heme Fe2+-chelate and ferric-chelate reductase
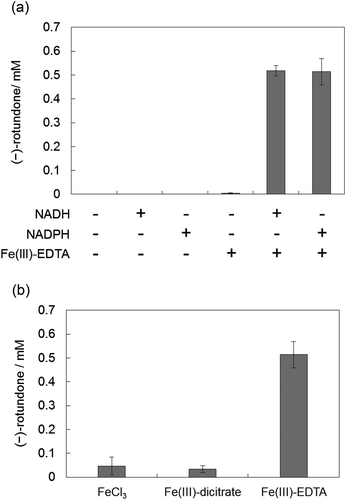
Fe2+-chelate supply and regeneration by YqjH was applied to (−)-rotundone synthesis. We selected FeCl3 as non-chelated iron, Fe(III)-dicitrate and Fe(III)-EDTA as chelated iron, which were previously reported to be reduced by YqjH [Citation26,Citation27]. As a result, (−)-rotundone was significantly synthesized with NAD(P)H, Fe(III)-EDTA, and YqjH, which indicates that the catalytic system with non-heme Fe2+-chelate and ferric-chelate reductase functioned properly ()). YqjH could utilize by both NADH and NADPH to reduce Fe(III)-EDTA. And a little amount of NADPH was consumed with FeCl3, and a little amount of (−)-rotundone was detected () and Figure S2). This result indicates the substrate specificity of YqjH is restricted to chelated form ferric iron. On the other hand, NADPH was consumed with Fe(III)-dicitrate, indicating the generation of Fe(II)-dicitrate by YqjH despite a low (−)-rotundone productivity () and Figure S2). This result suggests that the ligand structure of Fe2+-chelate is critical for catalytic oxygenation.
Figure 3. Synthesis of (−)-rotundone using YqjH. (a) Reaction mixtures contained α-guaiene, YqjH, and each cofactor described with a + symbol at 5 mM NAD(P)H or 1 mM Fe(III)-EDTA. (b) Reaction mixtures contained α-guaiene, NADPH, YqjH, and 1 mM of each ferric iron. Data show the means ± standard deviations of three independent experiments.
Evaluation of the catalytic system with Fe(III)-EDTA and YqjH
This catalytic system is composed of the Fe2+-chelate supply and regeneration by YqjH and the oxygenation by the Fe2+-chelate catalyst. In this regard, the Fenton-type reaction which causes oxidizing reactions using ferrous iron has been well investigated [Citation28]. Since the Fenton reaction requires H2O2 as an oxygen donor, we evaluated this catalytic system in the presence of catalase. Moreover, non-heme Fe2+-chelate catalysts have been suggested to generate alkyl radical intermediates during the catalytic cycle of oxygenation, and a radical scavenger, galvinoxyl free radical, could inhibit this cycle [Citation19,Citation23,Citation29]. Thus, we also determined the effect of this scavenger. As shown in –), even though an excess of catalase did not affect this system, the galvinoxyl free radical quenched the whole reaction. These results indicate that this oxygenation requires not H2O2 but presumably O2 as an oxygen donor and that Fe(II)-EDTA functions as the Fe2+-chelate catalyst via the generation of radical intermediates.
Figure 4. GC-MS analysis of the reaction products containing α-guaiene, NADPH, Fe(III)-EDTA, and YqjH (a), with 100 U/mL catalase (b), or 1 mM galvinoxyl free radical (c). MS spectra of each peak (d); α-guaiene (peak 1), (−)-rotundone (peak 2), and demethylated analog (peak 3).
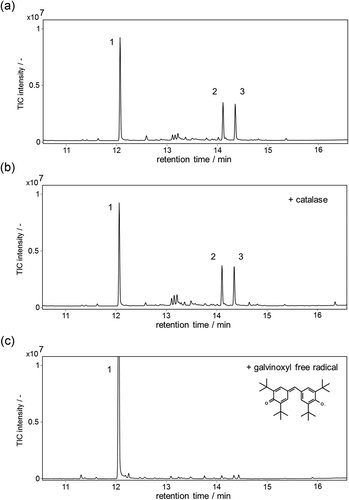
Figure 4. (Continued)
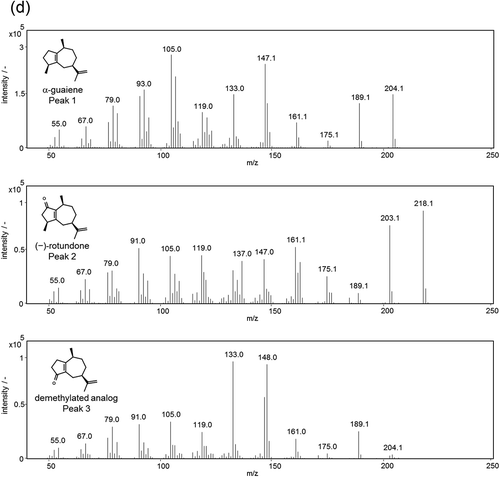
GC-MS analysis also showed that there existed a by-product with a similar retention time and of a similar amount to (−)-rotundone (, peak 3). According to the GC-MS fragment patterns and the UV absorption attributed to α,β-unsaturated carbonyl structure, we deduced that this by-product was an analog of (−)-rotundone. As such, this compound was isolated by silica gel column chromatography and preparative thin layer chromatography. Using 1H and 13C NMR spectroscopy, the structure was determined to be a demethylated analog of (−)-rotundone ()). This compound is a novel terpenoid, not currently registered on the CAS database, and has an earthy, musty odor that differs from (−)-rotundone. In addition, the odor profile of the product mixture which contained both (−)-rotundone and demethylated analog varied in accordance with those concentrations. The odor was earthy and musty at a high concentration around ppm order, and that was woody and peppery at a low concentration around ppt order. This profile indicates that there exist appropriate concentrations of these compounds for flavor and fragrance utilization.
Improvement of the efficiency of (−)-rotundone synthesis using cyclodextrin and glucose dehydrogenase
To improve the efficiency of (−)-rotundone synthesis, we addressed the stabilization of α-guaiene and the enhanced supply of NAD(P)H. The hydrophobicity and volatility of sesquiterpene often leads to low efficiencies in conversion reactions. Cyclodextrin is known to overcome this problem, and HP-β-CD, a hydroxypropylated derivative of β-cyclodextrin, is particularly suitable for sesquiterpene [Citation30]. The electron source of this catalytic system is NAD(P)H. The regeneration of these coenzymes could avoid the accumulation of NAD(P)+ and enhance the energy supply to facilitate the catalytic cycle. We selected glucose dehydrogenase from Bacillus sp., which was capable of reducing both NAD+ and NADP+.
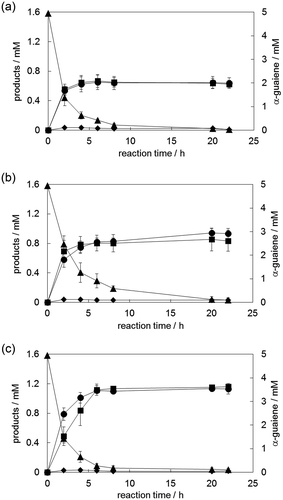
The addition of HP-β-CD to this system increased the amount of (−)-rotundone by 1.5-fold (,)). This effect was observed identically with both NADH and NADPH (Figure S3). Upon applying GlcDH to the (−)-rotundone synthesis, we optimized each parameter, such as GlcDH activity, and the types of coenzyme (i.e. NAD+ or NADP+). For an efficient catalytic cycle, 0.1 U/mL GlcDH and 1 mM of either NAD+ or NADP+ was sufficient (Figure S4). Then, we applied NADH regeneration with 0.1 U/mL GlcDH, 1 mM NAD+, and 100 mM glucose to the (−)-rotundone synthesis and found that the amount of (−)-rotundone improved to 1.2 mM, which corresponded to a 24% molar conversion rate against 5 mM α-guaiene ()). And this catalytic system functioned even when purified YqjH was replaced with recombinant cells expressing YqjH, and was scaled up to the flask size (Figure S5). Although the synthesis rate and yield of (−)-rotundone were slightly lowered compared to purified YqjH reaction, this would be due to membrane permeability of Fe2+-chelate or substrate and the margin of further improvement. Through these investigations, the demethylated by-product was generated at the same level as (−)-rotundone. The same amount of formaldehyde as demethylated by-products was also found to be generated (Figure S6). In addition to the structure of the by-product, this result also indicates that the Fe2+-chelate catalyst (this time Fe(II)-EDTA) possessed the catalytic activity for dissociating a methyl group from a substrate compound at an allylic position.
Figure 5. Time courses of (−)-rotundone synthesis using α-guaiene, NADPH, Fe(III)-EDTA, and YqjH (a), with 1% HP-β-CD (b) and 1 mM NAD+, 100 mM glucose, and 0.1 U/mL GlcDH as substitutes for NADPH (c). Circle, (-)-rotundone; square, demethylated analog; diamond, rotundol; triangle, α-guaiene. Data are shown as the means ± standard deviations of three independent experiments.
Discussion
The structure of over 80,000 terpenoids has been elucidated, and various useful functions have been identified [Citation1–Citation3]. However, many of these terpenoids are found in limited amounts in natural sources, such that effective synthesis methods are required for industrial applications. Here, we discovered that (−)-rotundone could be synthesized from precursor α-guaiene using the reduced form ferrous iron. Furthermore, we developed a unique catalytic system using non-heme Fe2+-chelate and ferric-chelate reductase, and we improved this system with the addition of glucose dehydrogenase and cyclodextrin. It is worth noting that this system is not restricted to (−)-rotundone synthesis but can also be applied to the synthesis of other terpenoids. Allylic oxidation could be applied to the synthesis of various terpenoids, and this research topic is currently being investigated. Moreover, another property of this catalytic system is that peroxide compounds are not required as an oxygen donor. This was confirmed through the use of catalase and galvinoxyl free radical, whereby we estimated that molecular oxygen functioned as an oxygen donor. Molecular oxygen is the most abundant natural oxygen donor, and the utilization of this donor plays a significant role in sustainable chemistry. Moreover, the application of GlcDH and HP-β-CD allowed for this catalytic cycle to be carried out without the costly addition of NAD(P)H and with a catalytic amount of NAD(P)+ and Fe3+-chelate.
On the other hand, the yield of (−)-rotundone remained at a molar conversion rate of 24% compared to α-guaiene. This low productivity could result from the generation of demethylated analog; however, the total (−)-rotundone and demethylated analog produced was approximately 2.5 mM, equal to 50% of the α-guaiene consumed. The remaining 50% was not traceable with either GC-MS or HPLC. As shown in , this catalytic cycle was quenched with a galvinoxyl free radical, which indicated the formation of alkyl radical intermediates during the oxidative reaction. Therefore, the condensation between radical-activated α-guaiene and another component of the reaction mixture, or among activated α-guaiene, may have caused the loss of substrate. The exclusion of unintentional loss of substrate is necessary for the further improvement of this method. In this regard, a more efficient Fe2+-chelate catalyst should be found to improve the efficiency of oxygenation. As shown in , the synthesis of (−)-rotundone was dependent on the ligand structure of Fe2+-chelate, which suggests that the optimization of the ligand could lead to a higher efficiency. Bleomycin, a glycopeptide-type anticancer drug isolated from Streptomyces verticillus, is a natural potent non-heme Fe2+-chelate catalyst [Citation31,Citation32]. Bleomycin possesses the activity to activate molecular oxygen and oxidatively cleave single and double strands of DNA with an intramolecular Fe2+-chelate structure which consists of penta-coordinate nitrogen ligands. It has been reported that N-ligand Fe2+-chelate catalysts, which are bleomycin mimics, also exhibit a high efficiency in oxygenation [Citation19,Citation20,Citation33]. Therefore, we infer that the yield of (−)-rotundone could be improved with those N-ligand Fe2+-chelate catalysts. This is the potential of this catalytic system to select the type of Fe2+-chelate catalysts according to objective reactions. Regarding the structure of demethylated analog of (−)-rotundone, several studies exist on the O-demethylation [Citation34], N-demethylation [Citation22,Citation35], or C-demethylation coupled with decarboxylase [Citation36] by non-heme iron enzymes or non-heme Fe2+-chelate catalysts. However, to the best of our knowledge, research on successive C-demethylation and carbonylation is uncommon. Further investigations on the reaction mechanism will be necessary.
Author contributions
K.K. and S.U. designed the experiment; S.U. and S.K. performed the experiments and analyzed the data; S.U. and K.K. wrote the manuscript.
Final_YqjH_Suppl._Figs.pptx
Download MS Power Point (615.9 KB)Acknowledgments
This research was supported in part by a Waseda University Grant for Special Research Projects (project number 2016A-032).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Related Research Data
References
- Gershenzon J, Dudareva N. The function of terpene natural products in the natural world. Nature Chem Biol. 2007;3:408–414.
- Schempp FM, Drummond L, Buchhaupt M, et al. Microbial cell factories for the production of terpenoid flavor and fragrance compounds. J Agric Food Chem. 2018;66:2247–2258.
- Bian G, Deng Z, Liu T. Strategies for terpenoid overproduction and new terpenoid discovery. Curr Opin Biotechnol. 2017;48:234–241.
- Wood C, Siebert TE, Parker M, et al. From wine to pepper: rotundone, an obscure sesquiterpene, is a potent spicy aroma compound. J Agric Food Chem. 2008;56:3738–3744.
- Cincotta F, Verzera A, Tripodi G, et al. Determination of sesquiterpenes in wines by HS-SPME coupled with GC-MS. Chromatography. 2015;2:410–421.
- Nakanishi A, Fukushima Y, Miyazawa N, et al. Identification of rotundone as a potent odor-active compound of several kinds of fruits. J Agric Food Chem. 2017;65:4464–4471.
- Nakanishi A, Fukushima Y, Miyazawa N, et al. Quantitation of rotundone in grapefruit (Citrus paradisi) peel and juice by stable isotope dilution assay. J Agric Food Chem. 2017;65:5026–5033.
- Takase H, Sasaki K, Shinmori H, et al. Cytochrome P450 CYP71BE5 in grapevine (Vitis vinifera) catalyzes the formation of the spicy aroma compound (−)-rotundone. J Exp Bot. 2016;67:787–798.
- Drew DP, Andersen TB, Sweetman C, et al. Two key polymorphisms in a newly discovered allele of the Vitis vinifera TPS24 gene are responsible for the production of the rotundone precursor α-guaiene. J Exp Bot. 2016;67:799–808.
- Sundaresan V, Singh SP, Mishra AN, et al. Composition and comparison of essential oils of Pogostemon cablin (Blanco) Benth. (Patchouli) and Pogostemon travancoricus Bedd. var. travancoricus. J Essent Oil Res. 2009;21:220–222.
- Tissandié L, Gaysinski M, Brévard H, et al. Revisiting the chemistry of guaiacwood oil: identification and formation pathways of 5,11- and 10,11-epoxyguaianes. J Nat Prod. 2017;80:526–537.
- Muzart J. Homogeneous CrVI-catalyzed benzylic, allylic and propargylic oxidations by tert-butyl hydroperoxide. Mini-Rev Org Chem. 2009;6:9–20.
- Felipe LO, Oliveira AM, Bicas JL. Bioaromas - Perspectives for sustainable development. Trends Food Sci Tech. 2017;62:153–414.
- Sales A, Paulino BN, Pastore GM, et al. Biogeneration of aroma compounds. Curr Opin Food Sci. 2018;19:77–84.
- Román S, Sánchez-Siles LM, Siegrist M. The importance of food naturalness for consumers: results of a systematic review. Trends Food Sci Tech. 2017;67:44–57.
- Janocha S, Schmitz D, Bernhardt R. Terpene hydroxylation with microbial cytochrome P450 monooxygenases. In: schrader J, Bohlmann J (eds) Biotechnology of Isoprenoids. Adv Biochem Eng Biotechnol. 2015;148:215–250.
- Schilling B, Granier T, Locher E, 1-Hydroxy-octahydroazulenes as fragrances. Patent WO2012/001018 A1. 2012
- Huang A-C, Burrett S, Sefton MA, et al. Production of the pepper aroma compound, (−)-rotundone, by aerial oxidation of α-guaiene. J Agric Food Chem. 2014;62:10809–10815.
- Hong S, Lee Y-M, Ray K, et al. Dioxygen activation chemistry by synthetic mononuclear nonheme iron, copper and chromium complexes. Coordin Cem Rev. 2017;334:25–42.
- Lindhorst AC, Haslinger S, Kühn FE. Molecular iron complexes as catalysts for selective C–H bond oxygenation reactions. Chem Commun. 2015;51:17193–17212.
- Abu-Omar MM, Loaiza A, Hontzeas N. Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem Rev. 2005;105:2227–2252.
- Nam W, Lee Y-M FS. Tuning reactivity and mechanism in oxidation reactions by mononuclear nonheme iron(IV)-oxo complexes. Acc Chem Res. 2014;47:1146–1154.
- Lee Y-M, Hong S, Morimoto Y, et al. Dioxygen activation by a non-heme iron(II) complex: formation of an iron(IV)-oxo complex via C-H activation by a putative iron(III)-superoxo species. J Am Chem Soc. 2010;132:10668–10670.
- Miethke M. Molecular strategies of microbial iron assimilation: from high-affinity complexes to cofactor assembly systems. Metallomics. 2013;5:15–28.
- Li K, Chen W-H, Bruner SD. Microbial siderophore-based iron assimilation and therapeutic applications. Biometals. 2016;29:377–388.
- Miethke M, Hou J, Marahiel MA. The siderophore-interacting protein YqjH acts as a ferric reductase in different iron assimilation pathways of Escherichia coli. Biochem. 2011;50:10951–10964.
- Wang S, Wu Y, Outten FW. Fur and the novel regulator YqjI control transcription of the ferric reductase gene yqjH in Escherichia coli. J Bacteriol. 2011;193:563–574.
- Barbusiński K. Fenton reaction - controversy concerning the chemistry. Ecol Chem Eng. 2009;16:347–358.
- Morimoto Y, Lee Y-M, Nam W, et al. An autocatalytic radical chain pathway in formation of an iron(IV)-oxo complex by oxidation of an iron(II) complex with dioxygen and isopropanol. Chem Commun. 2013;49:2500–2502.
- Lima PSS, Lucchese AM, Araújo-Filho HG, et al. Inclusion of terpenes in cyclodextrins: preparation, characterization and pharmacological approaches. Carbohyd Polym. 2016;151:965–987.
- Liu LV, Bell CB, Wong SD, et al. Definition of the intermediates and mechanism of the anticancer drug bleomycin using nuclear resonance vibrational spectroscopy and related methods. Proc Natl Acad Sci USA. 2010;107:22419–22424.
- Murray V, Chen JK, Chung LH. The interaction of the metallo-glycopeptide anti-tumour drug bleomycin with DNA. Int J Mol Sci. 2018;19:1372–1400.
- Geersing A, Ségaud N, van der Wijst MGP, et al. Importance of metal-ion exchange for the biological activity of coordination complexes of the biomimetic ligand N4Py. Inorg Chem. 2018;57:7748–7756.
- Hagel JM, Facchini PJ. Biochemistry and occurrence of O-demethylation in plant metabolism. Front Physiol. 2010;1:14.
- Yi C, Yang C-G, He C. A non-heme iron-mediated chemical demethylation in DNA and RNA. Acc Chem Res. 2009;42:519–529.
- Lee AK, Banta AB, Wei JH, et al. C-4 sterol demethylation enzymes distinguish bacterial and eukaryotic sterol synthesis. Proc Natl Acad Sci USA. 2018;115:5884–5889.