
ABSTRACT
The koji mold Aspergillus oryzae has been used in traditional Japanese food and beverage fermentation for over a thousand years. Amylolytic enzymes are important in sake fermentation, wherein production is induced by starch or malto-oligosaccharides. This inducible production requires at least two transcription activators, AmyR and MalR. Among amylolytic enzymes, glucoamylase GlaB is produced exclusively in solid-state culture and plays a critical role in sake fermentation owing to its contribution to glucose generation from starch. A recent study demonstrated that glaB gene expression is regulated by a novel transcription factor, FlbC, in addition to AmyR in solid-state culture. Amylolytic enzyme production is generally repressed by glucose due to carbon catabolite repression (CCR), which is mediated by the transcription factor CreA. Modifying CCR machinery, including CreA, can improve amylolytic enzyme production. This review focuses on the role of transcription factors in regulating A. oryzae amylolytic gene expression.
Graphical abstract
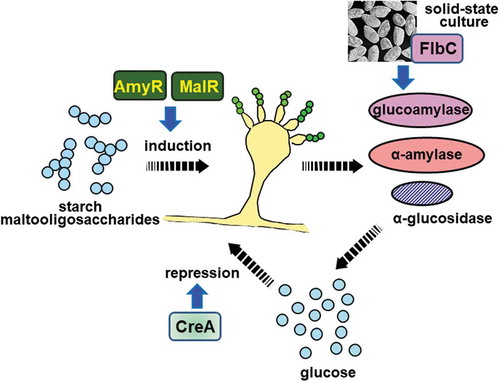
Aspergillus oryzae transcription factors involved in amylolytic enzyme production.
Washoku, which means traditional Japanese cuisine, was registered as a UNESCO Intangible Cultural Heritage in 2013 and gained more recognition worldwide. Washoku is constituted of traditional Japanese dishes, for which shoyu (soy sauce) and miso (soybean paste) are used as fundamental seasonings. In addition, the traditional Japanese alcoholic beverage, sake, is primarily paired with Japanese foods, such as sushi, tempura, and sashimi (fresh raw fish). Thus, shoyu, miso, and sake are significantly important fermented seasonings and beverages for washoku. The koji mold Aspergillus oryzae is indispensable for the production of fermented foods and has been used by the Japanese for over a thousand years, making it the most important filamentous fungus in Japan [Citation1,Citation2]. A. oryzae has even been heralded as the “national microorganism” of Japan [Citation3,Citation4].
A. oryzae produces copious amounts of hydrolytic enzymes required for fermentation. Amylolytic enzymes are essential for sake production, and proteolytic enzymes are essential for shoyu and miso production [Citation1,Citation2]. Amylolytic enzymes are comprised of α-amylase, which degrades starch by an endo-mechanism; glucoamylase, which hydrolyzes starch or dextrin by liberating glucose from the nonreducing end; and α-glucosidase, which primarily hydrolyzes maltose to glucose [Citation1]. The A. oryzae α-amylase is known as Taka-amylase A and has been molecularly characterized; its three-dimensional structure was elucidated by X-ray crystallography [Citation5] and its amino acid sequence [Citation6] determined. However, the α-amylase production mechanism has never been clarified until the development of a host-vector system that enabled genetic manipulation of A. oryzae [Citation7–Citation9]. There are previous reports describing α-amylase production in the presence of starch or malto-oligosaccharides, such as maltose or isomaltose, but not glucose [Citation10,Citation11]. We intended to elucidate the regulatory mechanisms for amylolytic enzyme production, including α-amylase, by employing our A. oryzae genetic engineering system. We expected that the regulatory mechanisms for amylolytic gene expression may be a suitable model to understand eukaryotic microbial gene expression and could be significantly useful for heterologous protein production by modifying the promoter activities of A. oryzae amylolytic genes. In this review, I describe the regulatory mechanisms of A. oryzae amylolytic gene expression, focusing on transcription factor function involved in amylolytic enzyme production.
The transcription factor AmyR is essential for the starch/maltose-inducible expression of amylolytic genes
Isolation of the gene encoding the transcription activator AmyR
To investigate the regulatory mechanisms of amylolytic gene expression, we first isolated A. oryzae genes encoding α-amylase (amyA/B/C) [Citation12], glucoamylase (glaA) [Citation13,Citation14], and α-glucosidase (agdA) [Citation15], all of which are induced by starch or maltose. Comparing the promoter region sequences of the three genes, we found three highly conserved sequences, designated as regions I, II, and III, other than TATA and CCAAT sequences, which are involved in gene expression [Citation16–Citation20]. Deletion analysis of these conserved sequences suggested that region III is mainly essential for high-level expression and maltose induction of amylolytic genes. We then expected that there is a transcription factor that can bind to region III; we aimed to isolate mutant strains deficient in α-amylase productivity to identify this gene via shotgun screening. However, it was difficult to isolate such a mutant strain because A. oryzae has multinuclear conidiospores, and another approach was necessary.
We constructed improved promoters by tandemly inserting multiple copies (up to 12 copies) of region III into the promoter region of the agdA and glaA genes [Citation21]. These promoters allowed hyperexpression of genes of interest ligated downstream of the promoters, indicating that the promoters can be used for high-level expression of heterologous genes [Citation21]. Concurrently, we observed that the amount of α-amylase produced was significantly reduced when expression constructs harboring multiple copies of region III was introduced in the transformants. As the number of integrated plasmid DNA increased, amyB gene expression decreased accordingly. This phenomenon seemed to be caused by the shortage of a transcription factor essential for amyB gene expression, as most of the factors may bind to multiple copies of region III, and only a small amount of it can bind to region III in the amyB promoter. If this is the case, we expected that using such a transformant as a host strain, a putative gene encoding the transcription factor could be isolated via screening the A. oryzae gene library. Consequently, we succeeded in isolating the transcription factor gene, which was designated as amyR [Citation22]. amyR mutants showed significantly poor growth on starch medium and also produced approximately 100-fold lower α-amylase and 10-fold lower glucoamylase levels compared with levels observed in the wild-type strain, demonstrating that AmyR is involved in amylolytic enzyme production. The deduced AmyR amino acid sequence showed the presence of a Zn(II)2Cys6 binuclear DNA-binding motif located close to the N-terminus [Citation22]. This motif was similar to the zinc finger motif of typical fungal transcription factors, such as Gal4 of Saccharomyces cerevisiae.
When we sequenced the isolated clone containing amyR, the 5′-flanking sequence of the agdA gene was located upstream of amyR. Based on this finding, we further sequenced the cosmid clones encompassing amyR and agdA and discovered one (amyA) of three identical α-amylase genes present in the A. oryzae genome downstream of the agdA gene. This indicated that amyR, agdA, and amyA constitute the amylolytic gene cluster, although another important amylolytic gene, glaA, did not exist in the cluster (). The amyR and agdA genes are transcribed divergently with an intergenic region of approximately 1.5 kb. However, different regulatory cis-elements seemed to exist in the intergenic region of these genes; northern blot analysis showed that agdA expression was induced by maltose but not glucose and that amyR was expressed irrespective of sugar species [Citation22]. Furthermore, interestingly, A. oryzae genome sequencing analysis [Citation23] revealed the existence of a transcription factor gene located adjacent to amyA (). This gene was shown to encode the transcription factor PrtR (PrtT) containing the Zn(II)2Cys6 binuclear DNA-binding motif and involved in proteolytic gene expression. Deletion of prtR resulted in a significant decrease in extracellular protease activity concomitant with a significantly reduced expression of proteolytic genes, including aspartic, neutral, and alkaline protease genes [Citation24]. prtR deletion also led to decreased peptidase gene expression (Matsuura et al., unpublished).
Figure 1. Schematic representation of the genomic region of the amylolytic gene cluster of A. oryzae and A. nidulans.
The gene sizes and locations were obtained from the Aspergillus genome database (AspGD; http://www.aspergillusgenome.org/). Arrows represent ORFs, showing transcriptional direction.
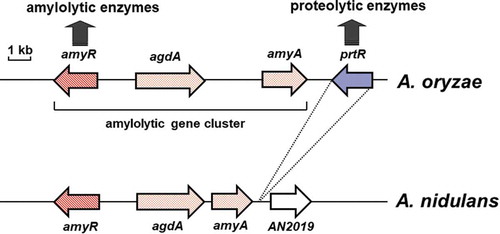
As described above, the transcription factors AmyR and PrtR are involved in amylolytic and proteolytic gene expression, respectively. Considering that amylolytic enzymes are essential for sake manufacturing, proteolytic enzymes are essential for shoyu and miso production, and the genome size and the number of the genes in A. oryzae are approximately 38 Mb and over 12,000, respectively [Citation23], it would be interesting to determine if the transcription factors involved in amylolytic and proteolytic gene expression are located in close proximity. Such a genome structure is present in other Aspergillus species, such as Aspergillus niger [Citation25] and Aspergillus fumigatus [Citation26]; however, Aspergillus nidulans does not have a PrtR ortholog [Citation25]. The amylolytic gene cluster also exists in the A. nidulans genome (), suggesting that regulatory mechanisms for proteolytic gene expression are evolutionarily differentiated among Aspergilli.
Sequence of the cis-element for AmyR binding
The AmyR amino acid sequence was shown to be highly homologous to that of the yeast Mal63, which is necessary for maltose utilization as it regulates genes encoding for the maltose transporter (Mal61) and maltase (Mal62; intracellular α-glucosidase) [Citation27,Citation28]. In particular, the similarity in the Zn(II)2Cys6 binuclear DNA-binding motif between AmyR and Mal63 suggested that AmyR may bind to a sequence similar to the Mal63-binding site. Mal63 is reported to bind to CGGN9CGG, CGCN9CGC, or CGGN9CGC in the intergenic regions of MAL61 and MAL62 or the MAL63 promoter [Citation29]. Because similar sequences are found in the conserved region III of A. oryzae amylolytic gene promoters (CGGN8AGG in amyB, CGGN8CCT in glaA, and CGG8CGG in agdA), AmyR likely binds to these sequences. In fact, a DNase-I foot printing assay revealed AmyR bound to the CGG triplet sequence in region III of amyB and glaA and additionally to the CGGN8CGG sequence present upstream of glaA region III [Citation30]. Furthermore, A. nidulans AmyR was able to bind to each CGG triplet of the sequence CGGN8CGG in the A. nidulans agdA promoter as a monomer; however, mutations in either CGG triplet resulted in the loss of inducible expression of agdA, suggesting that binding of AmyR to both CGG triplets is necessary for inducible gene expression [Citation31]. AmyR could bind to AAG in region III of the amyB promoter in the presence of the upstream CGG triplet [Citation32]. These results suggested that the binding site of AmyR may be CGGN8(C/A)GG. Furthermore, high-throughput screening procedure using the phage display system assisted with flow cytometry analysis demonstrated the AmyR-binding motif as CGGnnnTTTnTCGG [Citation33]. AmyR contains a leucine zipper-like heptad repeat sequence in the middle of the protein, and thus, AmyR likely binds to the region III sequence as a homodimer [Citation22].
The transcription factor MalR is involved in maltose utilization and amylolytic gene expression
Discovery of the maltose-utilizing (MAL) cluster from A. oryzae
As describe in Section I, disruption of the amyR gene resulted in a significantly reduced productivity of amylolytic enzymes, including α-amylase, leading to poor growth on starch medium compared with the wild-type strain [Citation22]. Because the production of glucoamylase and α-glucosidase was decreased, we assumed that amyR mutants may also show a restricted growth on the medium containing maltose as the sole carbon source. However, unexpectedly, amyR mutants exhibited an indistinguishable growth from the wild type on maltose medium, suggesting the existence of another system for maltose utilization other than what was regulated by AmyR in A. oryzae. In this context, it should be noted that deletion of amyR resulted in a restricted growth on both starch and maltose media in A. nidulans. An A. nidulans strain harboring malA mutation showed a growth defect on maltose medium, and this defect was recovered by the introduction of the wild-type amyR gene, meaning that AmyR would be also involved in maltose assimilation in A. nidulans [Citation34], which is different from the case in A. oryzae. Sequencing of the amyR gene from the malA mutant revealed a point mutation leading to substitution of histidine to leucine residue [Citation34].
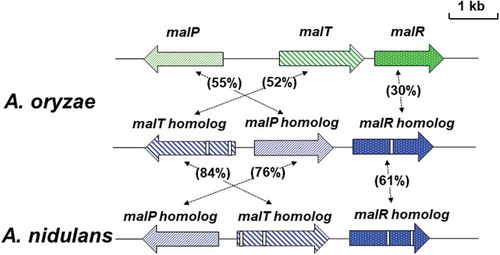
As mentioned previously, maltose utilization in the yeast requires the maltose-utilizing gene cluster (MAL cluster) composed of three genes that code for maltose transporter (Mal61), maltase (Mal62; intracellular α-glucosidase), and a transcription factor (Mal63) that regulates the expression of MAL61 and MAL62 [Citation27]. Analogously, we therefore presumed that a maltose utilization system similar to that in the yeast exists in A. oryzae, and a cDNA clone homologous to the MAL62 gene was found in the A. oryzae EST database [Citation35]. The nucleotide sequence of the cDNA clone containing the entire coding region revealed that the protein encoded by the cDNA showed high homology (46%) to the yeast Mal62, indicating that the cDNA encodes maltase or α-glucosidase, which is probably involved in maltose utilization in A. oryzae. Then, a genomic DNA clone was isolated by screening of approximately 10,000 clones of the phage library based on EMBL3 with the MAL62 homolog as a probe. This genomic DNA clone harbored an entire MAL61 homolog and a part of MAL63 homolog as well as the MAL62 homolog, suggesting the existence of a maltose utilization gene cluster similar to the yeast MAL cluster in the A. oryzae genome. According to the A. oryzae genome sequence database, we confirmed the gene structure of the maltose utilization gene cluster that consisted of the MAL61 homolog (designated as malP), MAL62 homolog (designated as malT), and MAL63 homolog (designated as malR) (). Gene organization of the cluster well resembles to the yeast MAL locus, although the malR gene is located downstream of the malT gene but not upstream of the malP gene, and thus, the gene cluster identified was designated as the MAL cluster in A. oryzae [Citation36].
Figure 2. Schematic representation of the MAL cluster and MAL homolog clusters of A. oryzae and A. nidulans.
The gene sizes and locations were obtained from the Aspergillus genome database (AspGD). The arrows represent ORFs, showing transcriptional direction. The open boxes in genes of the MAL homolog clusters indicate intron sequences. Amino acid sequence homologies are indicated.
The malP gene encodes a transmembrane protein of 539 amino acid residues with 12 putative transmembrane-spanning domains. Expression of the malP gene restored the growth on maltose medium of the yeast mutant deficient in maltose transporter [Citation37], indicating that MalP has a capability of transporting maltose across the plasma membrane. The hydropathy plot of MalT suggested the absence of a putative signal peptide sequence, indicating that MalT would be an intracellular α-glucosidase. When the malT gene was expressed under control of the high-level expression promoter in A. oryzae, a significantly high α-glucosidase activity was detected in the cell-free extract of the transformant, indicating that malT encodes a functional intracellular α-glucosidase. Although the malR gene could not complement the defect in mal63 mutation in the yeast, disruption of malR caused the loss of the expression of both malP and malT, demonstrating that the malR gene encodes the transcription factor required for malP and malT expression in A. oryzae [Citation36]. Furthermore, the expression of malP and malT was induced by maltose, but not glucose nor glycerol, and transcription of both genes was regulated by carbon catabolite repression because glucose addition resulted in the significant decrease in mRNA levels in maltose medium. By contrast, the malR gene was constitutively expressed with all carbon sources. This suggested that MalR is involved in maltose-inducible expression of malP and malT by MalR possibly following MalR activation in the presence of maltose. This hypothesis was drawn by the observation that malP deletion resulted in the loss of malT expression, provided that MalR could not be activated due to the absence of maltose incorporated by malP deletion. Additionally, deletion of amyR did not affect the malP and malT expression, confirming that maltose utilization was not regulated by AmyR [Citation36].
The MAL cluster involved in maltose utilization and amylolytic enzyme production
Deletion of each gene constituting of the MAL cluster genes resulted in poor growth on the medium containing maltose as the sole carbon source. (In the earliest experiment, malT disruption had no apparent effect on the growth on maltose medium [Citation36]. However, integrative gene mutations for malT used in the earliest study were unstable, and a deletion strain for malT newly constructed also showed a growth defect on maltose-containing medium.) This indicated that maltose utilization of A. oryzae would be attributed to the function of the MAL cluster. In addition to growth defects on maltose medium, deletion mutants of the MAL cluster genes also showed significantly poor growth on starch-containing medium. This suggested that starch utilization or amylolytic enzyme production may require the MAL cluster genes. Therefore, we examined α-amylase activities in the deletion mutants of each MAL cluster gene to elucidate the involvement of the MAL cluster in the amylolytic enzyme production.
The wild-type and deletion mutant strains were pre-cultured in the medium containing casamino acid and were then transferred to fresh maltose medium. Deletion of malR and malP genes resulted in a significant delay in the induction of α-amylase production; particularly, deletion of malP caused a severe defect in α-amylase production comparable to amyR deletion (). In addition to measuring α-amylase activities, the amount of maltose in the culture broth was determined so that maltose uptake by the strains can be estimated. Maltose in the culture broth was incorporated rapidly and completely consumed by the wild-type strain after incubation for 4 h. In contrast, the uptake of maltose by the malR deletion mutant was delayed by 4 h after transfer to maltose medium, whereas that by the malP deletion mutant was not observed until 8 h for incubation () [Citation38]. Considerable delay in maltose uptake by the malP mutant indicated that MalP was a major transporter required for maltose incorporation in A. oryzae. Furthermore, preliminary experiment performed recently showed that almost no consumption of maltose in the culture broth was observed in the malP deletion mutant in the presence of castanospermine, an inhibitor for extracellular α-glucosidase, suggesting that the decrease in the amount of maltose in the culture broth for 12-h incubation would be due to the degradation of maltose by extracellular α-glucosidase [Zan et al., unpublished]. This also supports that MalP would function as the sole maltose transporter present in A. oryzae, although the possibility cannot be ruled out that a certain sugar transporter(s) having a low affinity for maltose exists in A. oryzae.
Figure 3. Maltose consumption and α-amylase production in the wild-type and deletion mutant strains.
Each strain was grown in minimal medium containing 1% casamino acid as the carbon source for 24 h, followed by transfer to fresh medium containing 0.1% maltose. The amount of maltose (solid line) and α-amylase activity (dashed line) in the culture medium was determined at the time points indicated. The wild-type, malR, malP, and amyR deletion strains are represented by a diamond, square, triangle, and circle, respectively. The amounts and activities indicated in the panels are shown as the mean ± standard deviation of three independent experiments.
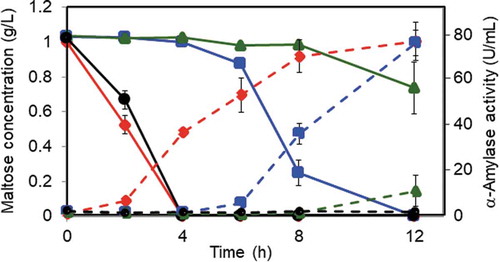
On the contrary, modest retardation of maltose uptake by the malR mutant suggested that the expression of malP was not completely lost by the malR deletion, and MalP produced from a small amount of its mRNA transcribed at the basal level was involved in maltose uptake at the later stage of incubation. Interestingly, maltose uptake patterns were closely related to the production of α-amylase in the malR and malP mutants, suggesting that maltose uptake by the maltose transporter MalP is necessary for α-amylase production in A. oryzae ().
According to the A. oryzae genome database, there was a paralogous gene cluster similar to the MAL cluster. This cluster is also comprised of three genes, all of which have high homology to genes in the MAL cluster: (a) malP homolog encoding a putative maltose transporter highly homologous (52% identity) to MalP; (b) malT homolog encoding a putative intracellular α-glucosidase homologous to MalT (55% identity); and (c) malR homolog encoding a putative transcription factor homologous to MalR (30% identity) () [Citation36]. Thus, the cluster was designated as the MAL homolog cluster. Although all three genes in the MAL cluster had no intron sequence, malT homolog and malR homolog genes except for malP homolog contained intron sequence. Despite the similarity of the gene organization and homology between the MAL cluster and MAL homolog cluster, however, no detectable signals derived from the MAL homolog cluster genes were observed by northern blot analysis even in the presence of starch or maltose, implying that the MAL homolog cluster genes were hardly expressed and thus were not involved in maltose utilization in A. oryzae [Citation36].
Amylolytic gene regulation mechanism required for both AmyR and MalR
Inducing sugar required for the activation of AmyR
As described above, we elucidated that the inducible production of amylolytic enzymes in A. oryzae is regulated by at least two Zn(II)2Cys6-type transcription factors, namely, AmyR and MalR. MalR is also essential for maltose utilization as it regulates the maltose transporter gene malP and the intracellular α-glucosidase gene malT [Citation36]. Although both amyR and malR genes are constitutively expressed [Citation22,Citation36], AmyR and MalR would be transcriptionally inactive in the absence of inducers, such as maltose, and would be activated by the addition of inducers. The activation mechanism of A. nidulans AmyR has been well studied, and isomaltose was proposed to be a real inducing sugar or physiological inducer for AmyR activation [Citation39,Citation40]. Amylase production in A. nidulans was examined by the addition of various α-linked disaccharides in the medium, and isomaltose (α-1,6-linked) could induce amylase production more significantly and at much lower concentration compared with other disaccharides, including maltose (α-1,4-linked), kojibiose (α-1,2-linked), and nigerose (α-1,3-linked). This was consistent with the previous observation that α-amylase induction efficiency of isomaltose was shown to be 1.8-fold higher than that of maltose in A. oryzae [Citation10]. The addition of the α-glucosidase inhibitor castanospermine clearly abrogated the α-amylase induction by maltose but, conversely, upregulated the induction by isomaltose. This result suggested that isomaltose converted from maltose by the transglycosylation activity of α-glucosidase is a physiological inducer for AmyR activation [Citation39]. Actually, in addition to the extracellular α-glucosidase AgdA, an ortholog of A. oryzae AgdA and A. niger AgdA, A. nidulans produced another extracellular α-glucosidase, AgdB, that showed a strong transglycosylation activity, which could play a major role in the conversion of maltose to isomaltose [Citation40]. Furthermore, it was reported in A. nidulans that AmyR was localized in the cytoplasm without inducing sugars and transported into the nucleus by the addition of inducers, including maltose and isomaltose [Citation41]. Both maltose and isomaltose could function as inducers for the nuclear localization of AmyR, but isomaltose could trigger AmyR nuclear import more rapidly and at a lower concentration compared with maltose [Citation42]. These observations also supported that isomaltose is the strongest and physiological inducing sugar for the A. nidulans AmyR activation.
According to the experiment results in A. nidulans, we examined the subcellular localization of AmyR as well as MalR in A. oryzae. As reported for A. nidulans AmyR, A. oryzae AmyR was localized in the cytoplasm under non-inducing conditions and translocated into the nucleus in the presence of maltose and isomaltose [Citation43]. Also, however, isomaltose triggered the nuclear import of A. oryzae AmyR more rapidly than maltose, implying that isomaltose is also a stronger inducer for A. oryzae AmyR activation. On the other hand, MalR was shown to be constitutively localized in the nucleus. Therefore, AmyR may be activated possibly by isomaltose and translocated from the cytoplasm to nucleus, but the nuclear localization of MalR is unrelated to its activation state [Citation43].
Conversion of maltose to isomaltose by the MAL cluster function
Unlike the case in A. nidulans, although maltose would be converted to isomaltose, a physiological inducer for A. oryzae AmyR activation, extracellular α-glucosidases seemed not to be involved in the conversion of maltose to isomaltose because the addition of α-glucosidase inhibitors, castanospermine and 1-deoxynojirimycin, could not inhibit the induction of α-amylase gene expression in the presence of maltose [Citation43]. This suggested that the conversion of maltose to isomaltose in A. oryzae occurs intracellularly. As described in Section II, α-amylase inducible production by maltose in A. oryzae requires the uptake of maltose into the cell by means of the MAL cluster (). Northern blot analysis showed that the α-amylase gene expression in the malR deletion mutant was normally induced by isomaltose but was scarcely observed after the addition of maltose [Citation43]. Similarly, the malP deletion resulted in the loss of α-amylase gene expression in the presence of maltose, although α-amylase induction took place normally by isomaltose [Ichikawa et al., unpublished]. Taken together with the observation that deletion of malT caused a severe induction delay in α-amylase production, all the members of the MAL cluster, MalR, MalP, and MalT, would be essential for the utilization of maltose as an inducer for AmyR activation. Among them, MalT would be the most plausible candidate required for the intracellular conversion of maltose to isomaltose. Thus, we examined the transglycosylation activity of the purified His-tagged MalT that was overexpressed in A. oryzae. As a result, MalT could form isomaltose and panose as well as glucose from maltose, indicating that MalT indeed had the transglycosylation activity, albeit to a lesser extent [Watanabe et al., unpublished].
MalR activation mechanism has not yet been clear because MalR was constitutively localized in the nucleus, but the expression of MAL cluster genes involved in maltose utilization was induced only by maltose, but not by isomaltose, indicating that maltose is the physiological inducer for MalR activation [Citation43]. Northern blot analysis showed that the induction of malT and malP gene expression by maltose was much earlier than that of the α-amylase gene expression, suggesting that MalR activation precedes AmyR activation in the presence of maltose as an inducer. It was possible that the lag time between α-amylase gene induction and malT or malP gene induction could be accounted for by the time required for the conversion of maltose to isomaltose by MalT. In contrast, when isomaltose was present as the inducing sugar, the expression of α-amylase gene was induced as rapidly as that of malT and malP genes in the presence of maltose because the conversion of maltose to isomaltose was not required [Citation43].
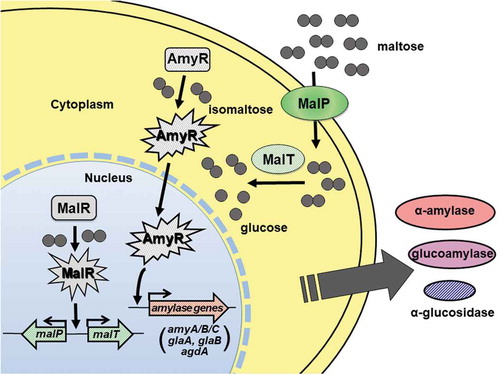
Altogether, on the basis of our experimental results, the regulation mechanism for amylolytic gene expression in the presence of maltose in A. oryzae was proposed as follows ():
A small amount of maltose is transported into the A. oryzae cell by the maltose transporter MalP whose gene is expressed at the basal level, which triggers the activation of the transcription factor MalR.
Transcription of malP and malT genes in the MAL cluster is rapidly induced by MalR activated by incorporated maltose, and then a large amount of maltose is transported into the cell by MalP.
The intracellular α-glucosidase MalT degrades maltose to glucose and concomitantly converts maltose to isomaltose by its transglycosylation activity, and resulting isomaltose triggers the activation and translocation of AmyR into the nucleus.
Amylolytic genes, such as amyA/B/C, glaA, glaB, and agdA, are induced by AmyR.
Figure 4. Schematic model for the induction of amylolytic gene expression mediated by MalR and AmyR. Detailed description of the model is referred to in the text.
Difference in amylolytic gene regulation between A. oryzae and A. nidulans
There are several differences in the regulation mechanisms of amylolytic gene expression between A. oryzae and A. nidulans. As described above, although isomaltose is in common the physiological inducer for AmyR activation, it is converted from maltose by intracellular α-glucosidase in A. oryzae but by extracellular α-glucosidase(s) in A. nidulans [Citation39]. The production of the intracellular α-glucosidase MalT that would be exclusively involved in the conversion of maltose to isomaltose is regulated by the transcription factor MalR, which is activated by maltose, in A. oryzae. By contrast, extracellular α-glucosidases, AgdA and AgdB mainly involved in isomaltose formation, could be induced by the transcription factor AmyR, which is activated by isomaltose, in A. nidulans [Citation39,Citation40]. Furthermore, no orthologous gene cluster of the MAL cluster exists in the A. nidulans genome, although there exists an orthologous cluster of the MAL homolog cluster () [Citation36,Citation44], which was hardly expressed under the conditions examined in A. oryzae as mentioned above [Citation36]. Expression profiles of the MAL homolog cluster in A. nidulans have not yet been studied in detail, but the expression of the agdF gene, an ortholog of malT homolog, was regulated by AmyR [Citation45]. Because A. nidulans is unable to efficiently incorporate maltose as judged from the observation that maltose in the culture broth was left unconsumed by the addition of the α-glucosidase inhibitor [Citation39], MalP homolog appears not to be sufficiently expressed or not to function as a maltose transporter in A. nidulans. This is clearly in contrast to the case in A. oryzae, where maltose could be rapidly imported by the function of MalP [Citation38]. The assumption that the MAL homolog cluster could not efficiently function in A. nidulans was supported by the observation that disruption of amyR in A. nidulans resulted in a growth defect not only on starch medium but also on maltose medium [Citation34], which indicated that maltose utilization of A. nidulans is governed by AmyR, but not MalR homolog. These observations clearly indicated that the regulatory mechanism for maltose utilization is different between A. oryzae and A. nidulans. Additionally, the mechanism of amylolytic enzyme production in A. oryzae also differs from that in A. nidulans, taking into account the involvement of the MAL cluster in the induction of α-amylase gene expression. Although both A. oryzae and A. nidulans belong to the same genus Aspergillus, it would be quite interesting from the point of view of fungal evolution that the regulatory mechanisms of amylolytic enzyme production differ between the two fungal species.
Chaperone complex involved in MalR activation
As described in Section II, the gene organization of the A. oryzae MAL cluster resembles that of the MAL cluster of the yeast. The transcription factor Mal63, the yeast counterpart of MalR, was reported to be transcriptionally activated by sequential complex formation with the chaperones Hsp70 and Hsp90 and the co-chaperone Sti1 in the presence of the inducing sugar maltose [Citation46,Citation47]. Taking account of the similarity between Mal63 and MalR, the inducer-mediated activation of MalR could analogously occur through the formation of a complex with such chaperone proteins. If this is applicable to MalR, the corresponding complex formation with chaperones may occur in the nucleus because MalR was constitutively localized in the nucleus. Co-immunoprecipitation (Co-IP) analyses using the co-expression strains of FLAG-tagged MalR with c-Myc-tagged Hsp70 or Hsp90 of A. oryzae showed that MalR formed a stable complex with both Hsp70 and Hsp90 under non-inducing conditions [Citation48]. Following the addition of the inducing sugar maltose, the chaperone Hsp90 was liberated first from the complex followed by Hsp70, suggesting that MalR is inactive which forms a complex with the chaperones Hsp70 and Hsp90 and becomes transcriptionally active by its release from the chaperones Hsp70 and Hsp90 in the presence of maltose [Citation48]. The importance of the temporal order in the sequential release of the chaperones in the MalR activation pathway has not yet been evident, but the activation of MalR obviously requires its release from the chaperone complex. Although MalR forms a complex with Hsp70 and Hsp90, the interaction mode of MalR with the chaperones appears to differ from that with Mal63. Mal63 forms a stable complex with Hsp70, Hsp90, and Sti1 under non-inducing conditions, which is similar to MalR. However, Hsp70 and Sti1 are released from the chaperone complex in the presence of the inducing sugar maltose, and the resulting complex composed of Mal63 and Hsp90 is an active form with DNA binding activity [Citation46,Citation47]. In contrast, the stable complex composed of MalR, Hsp70, and Hsp90 forms under non-inducing conditions, whereas Hsp90 is released first from the complex and then Hsp70 follows after the addition of maltose, and the resulting MalR probably free of the chaperones is transcriptionally active. The proteins associated with Hsp90 are generally destabilized by the loss of interaction with Hsp90 [Citation49,Citation50]; however, contrary to expectations, MalR showed to be stable even under inducing condition, where formation of MalR–Hsp90 complex was completely abolished [Citation48]. Furthermore, protein–protein interaction analysis using a MalR mutant defective in the nuclear localization showed the interaction manner with Hsp70 and Hsp90 chaperones was unaffected by its intracellular localization [Citation48].
Considering the similarity between MalR and AmyR, similar complex formation with the chaperones Hsp70 and Hsp90 could be also involved in the activation pathway of AmyR. Although detailed analysis has not been reported to date, a large protein complex comprising AmyR detected under non-inducing conditions was altered to a smaller size of complex in the presence of the inducer isomaltose in A. nidulans [Kobayashi, T., personal communication]. This suggested that similar complex formation with the chaperones occurs in the transcriptional activation accompanied with nuclear localization of AmyR. In contrast to MalR, AmyR appears to be unstable even under non-inducing conditions and is further destabilized by the addition of inducing sugars [Tanaka et al., unpublished].
The transcription factor FlbC required for extracellular hydrolases specifically produced in solid-state culture
Hydrolytic genes specifically produced in solid-state culture
The koji mold A. oryzae is generally cultivated in solid-state culture using rice grain, soybean, and wheat as the substrates for manufacturing the traditional Japanese fermented food, such as sake, shoyu, and miso [Citation1,Citation2]. One of the reasons why solid-state culture has been employed is that A. oryzae produces copious amounts of secreted hydrolytic enzymes, including amylolytic, proteolytic, and lipolytic enzymes required for the production of those fermented foods in solid-state culture compared with those in submerged culture. There have been many attempts for the development of high-level production systems of hydrolytic enzymes in submerged culture due to several disadvantages, such as difficulties in regulating culture conditions and the requirement of wider cultivation spaces in solid-state culture. However, all the attempts were ultimately unsuccessful due to the failure to obtain A. oryzae mutant strains with high-level productivity of amylolytic and proteolytic enzymes in submerged culture. The reason why such mutant strains could not be obtained has long remained to be elucidated. Recently, however, molecular genetic analysis in A. oryzae could open the way to solve the big question derived from the unsuccessful attempts.
As described in Section I, we have isolated three kinds of amylolytic genes, amyA/B/C, glaA, and agdA. Among them, the glucoamylase GlaA was highly homologous to A. niger GlaA and assumed to play a major role in liberating glucose in sake manufacturing [Citation13,Citation14]. However, several years later, another glucoamylase was purified especially from rice koji by the research group of Gekkeikan Co [Citation51]. This indicated that A. oryzae produced two kinds of glucoamylase, which showed differences in enzymatic properties, such as molecular mass and isoelectric point [Citation51]. From these results, glucoamylase purified from rice koji could be different from GlaA, and thereby, a novel gene encoding another glucoamylase (GlaB) was isolated [Citation52]. Quite interestingly, northern blot analysis showed that the mRNA corresponding to glaA was modestly expressed equivalently both in submerged and solid-state cultures, whereas a large amount of transcript for glaB was observed in the RNA preparation derived from solid-state culture, but not in that from submerged culture. This clearly indicated that the glaB gene is specifically expressed in solid-state culture, such as rice koji preparation [Citation52]. Deduced amino acid sequence of the novel glucoamylase GlaB revealed the lack of putative raw starch-binding domain, which exists in GlaA of A. oryzae and A. niger, suggesting that GlaB has no activity against raw starch. In the previous study, glucoamylase purified from rice koji showed no raw starch-digesting activity, well consistent with the prediction from the amino acid sequence of GlaB. Altogether, GlaB plays a crucial role in liberating glucose from starch, and its related malto-oligosaccharides essential for sake manufacturing and the glaB gene is expressed exclusively in solid-state culture. In addition to glaB, the pepA gene encoding aspartic protease [Citation53,Citation54] and the melB gene encoding tyrosinase [Citation55] of A. oryzae also showed to be specifically expressed in solid-state culture. Glucoamylase and proteases are required for sake and shoyu production, respectively, and these encoded genes are expressed exclusively in solid-state culture, but not in submerged culture, to which the failure of acquiring mutant strains with improved enzyme productivity can be attributed.
Identification of FlbC involved in hydrolytic genes specifically expressed in solid-state culture
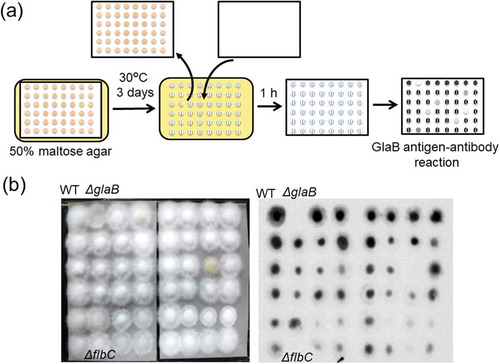
Since the glaB gene expression was lost by disruption of amyR, the maltose-induced expression of glaB is also regulated by AmyR [Citation56], but another unidentified transcription factor would be required for its specific expression in solid-state culture. Therefore, we attempted to identify such a transcription factor from the A. oryzae disruption mutant library for transcriptional regulators constructed by Noda Institute for Scientific Research [Citation57,Citation58] to screen the mutant strain that showed reduced GlaB production. It would be time-consuming and laborious to examine the GlaB production by cultivating mutants (over 400 strains) in solid-state culture. Growth conditions directing the glaB gene expression in solid-state culture can be mimicked by culture on the agar medium that fulfilled the reported inducing factors [Citation59]. Therefore, the mutant strains were grown on a nylon membrane with small pore size (physical barriers to hyphal extension) placed on the agar medium containing 50% maltose used as an inducer and to attain low water activity [Citation60]. After growth on the membrane, GlaB production was assessed by immunodetection using an anti-GlaB antibody (). The candidate strains screened showing the weak GlaB signals were grown on wheat bran as a substrate, and the glucoamylase activity in the crude enzyme extracts was determined. Consequently, one disruption strain showed a marked decrease in glucoamylase activity in solid-state culture compared with the wild-type strain [Citation60]. The strain isolated harbored disruption of the flbC gene encoding a C2H2-type transcription factor, which was reported to be involved in the regulation of conidiospore development in A. nidulans and A. oryzae [Citation61,Citation62]. Six upstream regulators, including FlbB, FlbD, and FlbE, are also involved in conidiospore development in addition to FlbC [Citation63], but the glucoamylase activities of all other disruption mutants were comparable with that of the wild-type strain, indicating that these upstream regulators were not involved in GlaB production and that FlbC was required for GlaB production in solid-state culture [Citation60].
Figure 5. Identification of the transcription factor required for GlaB production by screening the A. oryzae disruption mutant library.
(a) Schematic representation of the screening procedure. (b) An example of the screening results. Nylon membranes after A. oryzae growth and GlaB detection are presented in the left and right panels, respectively. Note that the flbC deletion mutant shows no apparent signal, similar to the glaB mutant.
As described above, expression of the aspartic protease gene pepA is also specifically induced in solid-state culture [Citation54], so we examined whether FlbC is involved in protease production. Protease activity under the acidic condition in the crude enzyme extract of the flbC mutant strain was markedly decreased, whereas those under neutral and alkaline conditions were comparable to those of the wild-type strain, suggesting that FlbC was also involved in aspartic protease production, but not in the neutral and alkaline protease production [Citation60]. Furthermore, the tyrosinase activity was not significantly altered between the flbC mutant and the wild-type strains. This result suggested that the influence of flbC disruption on tyrosinase production is negligible [Citation60].
Northern blot analysis revealed that expression of glaB and pepA were almost completely abolished by flbC deletion, although α-amylase gene transcripts were detected at a slightly lower expression level due to the growth defect of the flbC mutant. Besides glaB and pepA, interestingly, expression of the nptB gene encoding one of the major neutral proteases was also significantly downregulated in the flbC mutant. In contrast, transcripts of melB and the major alkaline protease gene alpA were not significantly affected by flbC deletion [Citation60]. These results clearly indicated that FlbC was involved in the regulation of glaB, pepA, and nptB gene expression in solid-state culture, although neutral protease activity was not influenced by flbC deletion. Furthermore, flbC complementation restored GlaB production, indicating that FlbC was indeed involved in the specific regulation of GlaB production in solid-state culture [Citation60]. Also, the transcription level of glaB, pepA, and nptB genes was restored by flbC complementation, but surprisingly, flbC deletion resulted in a significant increase in the transcription level of nptA encoding another major neutral protease, which was abolished by flbC complementation [Arai et al., unpublished]. This suggested that FlbC functions as a repressor for nptA gene expression and could account for why neutral protease activity was not influenced by flbC deletion, despite the loss of expression of nptB in the flbC mutant.
In conclusion, FlbC is involved in the expression of the genes, including glaB, pepA, and nptB, which are expressed specifically in solid-state culture. The next question is what sequence in the gene promoters is responsible for FlbC binding. The sequence TGACGA[T/A] was predicted by MEME sequence analysis of the promoter regions of brlA, abaA, and vosA to which FlbC can directly bind, and the sequence GTAGATC was predicted by another approach as the candidate for A. nidulans FlbC-binding site [Citation61], but it has not yet been demonstrated that FlbC actually binds to the sequences by electrophoretic gel mobility shift assay (EMSA). On the other hand, EMSA analysis using whole-cell extracts from A. oryzae indicated that there were three sequence elements predicted as the DNA-binding sites in the glaB promoter region [Citation64]. Among them, the substitution of the sequence element containing GC-rich sequence resulted in the complete loss of transcriptional activity of the glaB promoter in solid-state culture [Citation65], suggesting that such a GC-rich sequence is the candidate of the FlbC-binding site. However, our recent EMSA analysis using the FlbC protein heterologously produced by Escherichia coli preliminarily suggested that FlbC could recognize the region ranging between AmyR-binding motif (region III-like sequence) and TATA element as the binding site [Arai et al., unpublished]. Interestingly, the region contained the sequence element similar to GTAGATC sequence. Recent papers also reported that the FlbC orthologs of Neurospora crassa [Citation66] and Magnaporthe oryzae [Citation67] can bind to the similar sequence elements containing GATC sequence. These observations indicated that A. oryzae FlbC appears to bind to the region containing GATC sequence nearby TATA-box, but not to GC-rich sequence, to which another unidentified transcription factor might bind, in the glaB promoter.
Increased hydrolytic enzyme production by deletion of carbon catabolite repression machinery
Increased α-amylase production by double creA and creB deletion
The production of amylolytic enzymes in A. oryzae is induced by starch or its degradation intermediates, such as maltose and isomaltose, and in contrast, strongly repressed in the presence of glucose, the final product resulted from starch degradation. Repression of the gene expression initiated by glucose is caused by a well-known regulation mechanism called carbon catabolite repression (CCR). CCR in filamentous fungi is regulated directly by the C2H2-type transcription factor CreA [Citation68]. According to genetic and molecular analyses in A. nidulans, CreB and CreC have also been found to be regulating factors for CCR. CreB is a deubiquitinating enzyme that deubiquitinates CreA together with the WD40 repeat protein CreC to allow CreA activation [Citation69,Citation70]. Furthermore, ubiquitination of CreA is mediated by the ubiquitin ligase HulA with the assistance of the arrestin-like adaptor protein CreD [Citation71]. We assumed that similar regulatory mechanism works in the repression of amylolytic gene expression in A. oryzae and attempted to generate single- and double-deletion mutants for creA and creB to improve the production yields of secreted hydrolytic enzymes, including α-amylase, by the relief from CCR in A. oryzae [Citation72].
Whereas a creB deletion mutant grew normally comparable to the wild-type strain on glucose agar medium, single creA and creA/creB double mutants showed poor growth on the medium, consistent with the observations previously reported in other filamentous fungi [Citation68,Citation73]. However, in submerged culture using YPD medium (yeast extract, polypeptone, and glucose), creA deletion mutants showed a slightly lower growth at the first day of cultivation, but at the second day, their mycelial weights exceeded those of the wild-type and creB mutant strains. Although poor growth phenotype of creA deletion mutants has been regarded as a disadvantage for industrial enzyme production, our results indicated that creA mutants of A. oryzae could grow better than the wild-type strain in submerged culture, particularly for a longer cultivation period, in nutrient-rich medium, and thus, creA deletion appeared not to be disadvantageous in industrial use. More intriguingly, creA deletion mutants grew in significantly small pellets or with a pulpy-like morphology, whereas both the wild-type and creB mutant strains generally grew in a typical compact mycelial pellet. This distinctive morphological property of dispersed mycelial growth in creA deletion mutants likely accounts for increased mycelial weights at the second day [Citation72].
All creA and creB deletion mutants displayed a zone of clear halo surrounding the colonies on the agar media containing starch with glucose, in contrast to the wild-type strain that could form no clear zone around the colony, indicating that CCR was ameliorated by the deletion of either creA or creB and both. Then, the production of α-amylase in the mutants was examined in submerged culture. In YPD medium, the wild-type and creB mutant strains showed a low α-amylase activity, whereas the creA and creA/creB deletion mutants produced a large amount of α-amylase (). The observation that creA deletion resulted in a significant increase in α-amylase production yield suggested that glucose could act as an inducer for amylolytic enzyme production. This was supported by the facts that AmyR could be translocated in the nucleus even in the presence of glucose, albeit high concentration required in both A. oryzae and A. nidulans [Citation42,Citation43]. In YPM medium containing maltose, single creA or creB deletion mutant showed higher α-amylase activity compared with the wild-type strain, and creA/creB double-deletion mutant demonstrated the highest productivity of α-amylase. Particularly, the α-amylase activity of the double-deletion mutant in the medium containing 5% maltose or starch was more than tenfold higher than that of the wild-type strain () [Citation72]. These results suggested that deleting both creA and creB causes dramatic improvements in the α-amylase production and could lead to the anticipation that the productivities of other glycoside hydrolases are increased by creA and creB double deletion, which will be described below.
Figure 6. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of the creA and creB deletion mutants.
The wild-type and deletion mutant strains were grown in YP + 5% sugar (glucose or maltose) for 48 h at 30°C. Mycelia were incubated in 100 mM phosphate buffer (pH 7.0) for 1 h to release enzymes bound to the cell wall. The culture broth and phosphate buffer used for releasing enzymes from the cell wall were mixed and subjected to SDS-PAGE.
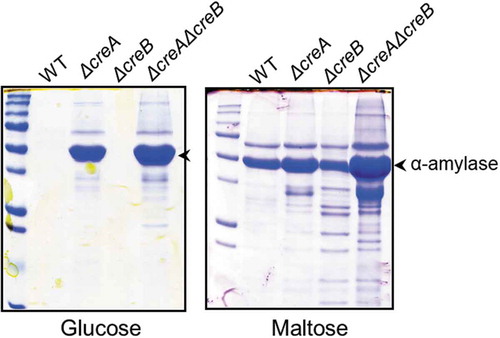
The mRNA levels of α-amylase gene in the creA and creA/creB mutants grown in maltose medium were markedly higher than that in the wild-type strain, indicating that creA deletion is considerably effective for enhancing the transcription level of the α-amylase gene and results in α-amylase production increase. However, although creB deletion increased the α-amylase productivity comparable to creA deletion, the expression level of the α-amylase gene in the creB mutant showed to be much lower than that in creA mutants [Citation72]. The reason is still unclear, but CreB is a deubiquitinating enzyme, and it is possible that there are several target proteins for CreB other than CreA, and target protein(s), as yet unidentified, might be involved in the repression of α-amylase production, presumably at the post-transcriptional level. This is also the case in which double-deletion mutant showed the highest α-amylase productivity, despite having no apparent difference in the transcription level of α-amylase gene between single creA and double-deletion mutants [Citation72]. Furthermore, the creA and double-deletion mutants showed more than twice as much α-amylase activity compared with the wild-type strain in solid-state culture using wheat bran as the substrate. This suggested that creA deletion also positively impacts α-amylase production in solid-state culture. In contrast to the case in submerged culture, no apparent effect on α-amylase production could be observed by creB deletion in solid-state culture [Citation72].
Increased biomass-degrading enzyme production by double creA and creB deletion
Filamentous fungi, including A. oryzae, are known to efficiently produce plant biomass-degrading enzymes, such as cellulases and xylanases. Production of these biomass-degrading enzymes is also repressed due to CCR. Therefore, it could be reasonably predicted that deletion of creA and creB is also effective for the improvement of biomass-degrading enzyme productivity [Citation74]. Interestingly, the growth of the wild-type and creB mutant strains in the liquid medium containing xylose as the carbon source to induce biomass-degrading enzyme production was significantly impaired in the presence of a high xylose concentration (5%), whereas creA deletion mutants apparently showed normal growth in such media [Citation74]. The transcriptional level of the gene encoding the transcription factor XlnR was also distinctly decreased in the wild-type and creB mutant strains compared with that of creA deletion mutants. The results suggested that a higher xylose concentration represses the xlnR gene expression, and creA deletion ameliorates the repression of xlnR expression, which promotes growth recovery [Citation74]. The expression of the genes encoding xylose catabolic enzymes, such as xylose reductase and xylitol dehydrogenase, was reported to be regulated by XlnR, which also regulated a putative xylose transporter gene expression [Citation75]. Thus, it is possible that, in the presence of a high xylose concentration, the expression level of xlnR was downregulated, which in turn resulted in the downregulation of the xylose transporter and catabolic genes, finally leading to reduced xylose uptake and assimilation.
In addition to the impact of creA deletion on the growth in high xylose medium, single creA and creA/creB double mutants showed a significantly increased productivity of xylanase and β-glucosidase. In contrast to the case of α-amylase, creB deletion did not significantly affect the productivity of xylanase and β-glucosidase. The expression levels of a xylanase gene, xynG2, and a β-glucosidase gene, bglC, were considerably increased by single creA and creA/creB double deletion. These results indicated that expression of the genes encoding xylanase and β-glucosidase are de-repressed by creA deletion, resulting in the hyperproduction of those enzymes [Citation74]. In conclusion, deletion of creA and creA/creB can considerably improve the production of biomass-degrading enzymes, such as xylanase and β-glucosidase, by the relief from CCR in A. oryzae. On the other hand, cellulase (endo-β-glucanase) activity was not affected by any deletions, suggesting that CCR in cellulase production is unrelated to CreA and CreB [Citation74]. This may be supported by the observation that cellulase production in the presence of cellulosic sugars with glucose was not de-repressed by the creA deletion, but the deletion of protein kinase A gene (pkaA) could ameliorate the CCR regulation in A. nidulans [Citation76,Citation77].
Mycelial morphology grown in submerged culture of the creA deletion mutant
As mentioned above, creA deletion mutants displayed an intriguing morphological phenotype of dispersed mycelial growth in submerged culture [Citation74]. The property of mycelial morphology is similar to that previously observed in the deletion mutants for the agsB gene encoding one of the three α-1,3-glucan synthases present in the A. oryzae genome [Citation78,Citation79]. The deletion of other α-1,3-glucan synthase genes, agsA and agsC, had no apparent impact on the mycelial morphology grown in submerged culture, and the agsB mutant mostly lost α-1,3-glucan in the cell wall, indicating that AgsB plays a critical role in α-1,3-glucan synthesis in A. oryzae [Citation79]. Similar mycelial growth phenotype was also observed in the agsB mutants in A. nidulans [Citation80]. Since α-1,3-glucan in the cell wall was suggested to promote aggregation of the mycelium [Citation81], the loss of α-1,3-glucan may result in mycelium dispersion in submerged culture by preventing mycelial adhesion, leading to the generation of pulpy-like morphology.
Morphological similarity between creA and agsB deletion mutants implies that creA deletion might downregulate the expression of the agsB gene resulting in the α-1,3-glucan content decrease in the cell wall. Contrary to expectation, transcriptional level of agsB was inversely upregulated by creA deletion [Ichinose et al., unpublished]. In A. nidulans, amyG and amyD, which form a gene cluster together with agsB, are suggested to be involved in the synthesis and degradation of α-1,3-glucan, respectively [Citation82,Citation83]. This gene cluster also exists in the A. oryzae genome, and the impact of creA deletion on the expression of amyG and amyD genes was examined. Whereas the amyG expression level was not significantly altered by creA deletion, amyD gene expression was considerably upregulated in the creA mutant [Ichinose et al., unpublished]. These observations that both agsB and amyD genes were upregulated by creA deletion suggested that the synthesis and degradation of α-1,3-glucan are simultaneously facilitated in the creA mutant. Overexpression of the amyD gene caused a decrease in the amount of α-1,3-glucan in the cell wall in A. nidulans [Citation82,Citation83]. This result implies that the amount of α-1,3-glucan synthesized by AgsB is decreased by the increased AmyD activity in the creA mutant, leading to the dispersion of the mycelium. Alternatively, it is possible that the molecular mass of the cell wall α-1,3-glucan synthesized by AgsB is lowered by the AmyD activity, leading to reduced adhesion strength of the cell wall, based on the observation that smaller mycelial pellets were generated due to the smaller α-1,3-glucan formed by amyG deletion in A. nidulans [Citation84]. Further analysis is required to clarify the molecular mechanism by which the creA deletion mutant showed a dispersed mycelial growth, but the resulting mycelial morphology seems favorable for the increase of mycelial weights and also for the improvement of the production yields of secreted hydrolytic enzymes in submerged culture, as reported for the agsB deletion mutant in A. oryzae [Citation78].
Future perspectives
The regulatory mechanisms for amylolytic gene expression have been mostly elucidated by our studies as illustrated in . However, some issues still remain unsolved, the most important issue being the limited understanding of how isomaltose and maltose induce AmyR and MalR activation, respectively. The addition of the inducing sugars led to the release of AmyR or MalR from the chaperone complex [Citation48]. The deletion of the C-terminal region of the A. nidulans AmyR protein led to constitutive activation of α-amylase gene expression [Citation41], suggesting that the C-terminal region of AmyR may act as a repressor domain under a non-inducing condition. Hence, isomaltose might induce a conformational change in AmyR directly or indirectly, resulting in its release from the chaperone complex and transcriptional activation. Similar mechanism is predicted in the case of MalR and maltose, and future studies are required to elucidate the activation mechanisms of AmyR and MalR.
The wild-type strain of A. oryzae can efficiently incorporate maltose by the function of the maltose transporter MalP [Citation38], but our preliminary study implies that isomaltose could be barely transported into the cell [Ichikawa et al., unpublished]. This finding is similar to the case in A. nidulans, where both maltose and isomaltose seemed to be unconsumed in the presence of the α-glucosidase inhibitor [Citation39]. Considering that an extremely low concentration of isomaltose could induce α-amylase production and the nuclear localization of AmyR in A. nidulans [Citation42], quite a small amount of isomaltose incorporated in the cell could be enough to transcriptionally activate AmyR. Therefore, it is possible that there is a certain sugar transporter(s) with a low affinity or a low transport activity for isomaltose that can import an undetectable amount of isomaltose. Alternatively, it would also be possible that a receptor or transporter-like sensing protein(s) for isomaltose exists in A. oryzae and A. nidulans. Recently, transporter-like proteins that also function as receptors for nutrient sensing, so-called transceptors, have been documented in yeasts and filamentous fungi [Citation85]. Transceptors are divided into two groups: membrane proteins that possess transporting and additional signal sensing activities and membrane proteins that have transporter-like structures but no transporter activity [Citation85,Citation86]. In filamentous fungi, N. crassa Cdt1/2 [Citation87] and Trichoderma reesei Stp1 [Citation88] possess cellodextrin or cellobiose transporter activity and cellobiose sensing activity, and T. reesei Crt1 [Citation88] and N. crassa Clp1 [Citation89] possess cellobiose sensing activity but no cellobiose transporter activity. These facts suggest that there might exist a putative isomaltose transceptor without transporter activity in A. oryzae and A. nidulans. However, if this is the case, a question of why A. oryzae must have two different pathways, namely, intracellular and extracellular, for sensing isomaltose for the activation of AmyR arises. Isolation of a putative transporter or transceptor for isomaltose should be required to answer the question.
In our preliminary study, we have found that the deletion of the MAL cluster genes caused upregulation of the MAL homolog cluster genes [Zan et al., unpublished], of which expression was undetectable even in the presence of starch or maltose in the wild-type strain [Citation36]. The observation suggested that A. oryzae could evolve a “fail-safe” mechanism to ensure the efficient utilization of starch in case of accidental loss of function of the MAL cluster, although the functionality of the MAL homolog cluster has not yet been fully elucidated.
Furthermore, from the evolution point of view, Aspergillus fungi seemingly possess the variety of regulatory mechanisms for amylolytic gene expression. As described above, the physiological inducing sugar isomaltose is converted from maltose intracellularly in A. oryzae but extracellularly in A. nidulans. This difference might arise from divergent evolution of the strategy to efficiently convert maltose to isomaltose. In this regard, the amylolytic gene regulation in A. luchuensis and A. niger, both belonging to Aspergillus section Nigri, should be investigated. A. luchuensis has been long used for shochu (Japanese spirits) manufacturing, mainly in southern areas of Japan, such as Okinawa and Kyushu islands. It produces a copious amount of amylolytic enzymes [Citation90,Citation91]. A. niger has been used for the industrial production of glucoamylase and citric acid [Citation92]. Interestingly, no MAL cluster is found in the A. luchuensis and A. niger genomes, although each corresponding gene with a low homology is dispersed in their genomes [Citation36,Citation44,Citation93]. Disruption of amyR in A. niger reduced productivities of secreted amylolytic enzymes, including glucoamylase (GlaA), α-glucosidases (AgdA and AgdB), and acid-stable α-amylase (AamA), implying the presence of AmyR-mediated regulatory mechanism in A. niger [Citation44,Citation94]. In A. luchuensis, the amyR deletion mutant showed significantly reduced expression levels of the glaA, agdA, and aamA genes [Hashimoto et al., unpublished]. However, acid-unstable α-amylase (AmyA), identical to A. oryzae α-amylase (TAA), was produced irrespective of sugar species [Citation95], and its gene expression was not influenced by the amyR deletion [Hashimoto et al., unpublished]. Furthermore, it was proposed in A. niger that extracellular maltose is degraded to glucose by glucoamylase and α-glucosidase, and the resulting glucose is taken up in the cell due to the absence of the MAL cluster [Citation44]. Hence, the conversion of maltose to isomaltose would be also catalyzed by extracellular α-glucosidase in A. niger and A. luchuensis, as reported in A. nidulans.
The C2H2-type transcription factor FlbC has been shown to be essential for the transcription of the genes specifically expressed in solid-state culture such as koji preparation [Citation60]. Further study should be required to elucidate the signaling pathways that transmit the signals from environmental stimuli generated when grown on solid substrates to the transcription factor FlbC. The glaB gene was particularly upregulated by growth under the conditions with decreased water availability and inhibition of hyphal extension [Citation59], causing environmental stresses on the cell. Thus, the study on the involvement of mitogen-activated protein (MAP) kinase pathways [Citation96] in this signal transduction system is underway. In addition, acid-stable α-amylase, AamA, of A. luchuensis was exclusively produced in solid-state culture [Citation95], and thus it would be interesting whether FlbC ortholog is involved in the aamA gene expression.
A double creA and creB deletion is highly effective for improving the production levels of extracellular hydrolytic enzymes, such as α-amylase, xylanase, and β-glucosidase [Citation72,Citation74]. Furthermore, we recently reported that the production yield of α-amylase was significantly elevated in the creB deletion mutant by dephosphorylated mutation in CreD, which is proposed to assist CreA ubiquitination by the ubiquitin ligase HulA [Citation97]. Therefore, modification of the function of the CCR machinery would be one of the promising procedures for increasing the production levels of secreted hydrolytic enzymes in filamentous fungi. However, although the observation that CreD was required for de-repression from CCR by creB deletion suggested the possible function of CreD as an adaptor of CreA ubiquitinated by HulA, CreD was not involved in CreA degradation because creD deletion had no effect on CreA stability. Similarly, CreA stability was not influenced by mutation or deletion of creB in A. nidulans or A. oryzae [Citation98,Citation99]. These results raised the question of whether or not CreA function is indeed directly regulated by ubiquitination/deubiquitination. Ubiquitination of CreA is deemed as a controversial issue due to the question as to whether ubiquitinated CreA was detected in A. nidulans [Citation98,Citation100]. The CreA protein was imported into the nucleus and stabilized in the presence of glucose and exported to the cytoplasm and destabilized in the presence of maltose or xylose, similar to the regulation mechanism by nuclear shuttling of the yeast counterpart Mig1. Although single creB deletion did not apparently affect the expression of α-amylase gene, deletion of creB in the creA mutant showed the additive effect on the productivities of hydrolytic enzymes, including α-amylase, as described above. These observations seemingly imply that CreA might have not been directly regulated by ubiquitination/deubiquitination mediated by CreB, CreC, CreD, and HulA, which might be involved in ubiquitination/deubiquitination of a CreA-associated protein(s) required for CreA function [Citation77,Citation101]. Further study on the regulation of CreA function could provide a better understanding of the detailed molecular mechanism of CreA-mediated CCR in filamentous fungi.
Acknowledgments
The studies described in this review were conducted at the National Research Institute of Brewing and at the Graduate School of Agricultural Science, Tohoku University. I am sincerely grateful to all of my colleagues who worked with me or assisted with my research. Particularly, I thank the late Dr. Hiroichi Akiyama, the former president of the National Research Institute of Brewing, for providing me with the opportunity to study A. oryzae, and I also thank Dr. Yuzuru Iimura, Professor Emeritus of the University of Yamanashi, for his collaboration in the development of an A. oryzae genetic engineering system. I am also thankful to Dr. Teruhiko Beppu, Professor Emeritus of the University of Tokyo, and the late Dr. Gakuzo Tamura, Professor Emeritus of the University of Tokyo, for their warm encouragement.
The author would like to thank Enago (www.enago.jp) for the English language review.
Disclosure statement
No potential conflict of interest was reported by the author.
Additional information
Funding
References
- Sakaguchi K, Takagi M, Horiuchi H, et al. Fungal enzymes used in oriental food and beverage industries. In: Kinghorn JR, Turner G, editors. Applied molecular genetics of filamentous fungi. Glasgow (UK): Blackie & Son; 1992. p. 54–99.
- Machida M, Yamada O, Gomi K. Genomics of Aspergillus oryzae: learning from the history of koji mold and exploration of its future. DNA Res. 2008;15:173–183.
- Kitamoto K. Cell biology of the koji mold Aspergillus oryzae. Biosci Biotechnol Biochem. 2015;79:863–869.
- Ichishima E. Development of enzyme technology for Aspergillus oryzae, A. sojae, and A. luchuensis, the national microorganisms of Japan. Biosci Biotechnol Biochem. 2016;80:1681–1692.
- Akabori S, Ikenaka T, Hagihara B. Isolation of crystalline taka-amylase A from “Takadiastase Sankyo”. J Biochem. 1954;41:577–582.
- Toda H, Kondo K, Narita K. The complete amino acid sequence of Taka-amylase A. Proc Jpn Acad Ser B. 1982;58:208–212.
- Iimura Y, Gomi K, Uzu H, et al. Transformation of Aspergillus oryzae through plasmid-mediated complementation of the methionine-auxotrophic mutation. Agric Biol Chem. 1987;51:323–328.
- Gomi K, Iimura Y, Hara S. Integrative transformation of Aspergillus oryzae with a plasmid containing the Aspergillus nidulans argB gene. Agric Biol Chem. 1987;51:2549–2555.
- Yamada O, Lee BR, Gomi K. Transformation system for Aspergillus oryzae with double auxortophic mutations, niaD and sC. Biosci Biotechnol Biochem. 1997;61:1367–1369.
- Tonomura K, Suzuki H, Nakamura N, et al. On the inducers of α-amylase formation in Aspergillus oryzae. Agric Biol Chem. 1961;25:1–6.
- Yabuki M, Ono N, Hoshino K, et al. Rapid induction of α-amylase by nongrowing mycelia of Aspergillus oryzae. Appl Environ Microbiol. 1977;34:1–6.
- Tada S, Iimura Y, Gomi K, et al. Cloning and nucleotide sequence of the genomic Taka-amylase A gene of Aspergillus oryzae. Agric Biol Chem. 1989;53:593–599.
- Hata Y, Kitamoto K, Gomi K, et al. The glucoamylase cDNA from Aspergillus oryzae: its cloning, nucleotide sequence, and expression in Saccharomyces cerevisiae. Agric Biol Chem. 1991;55:941–949.
- Hata Y, Tsuchiya K, Kitamoto K, et al. Nucleotide sequence and expression of the glucoamylase-encoding gene (glaA) from Aspergillus oryzae. Gene. 1992;108:145–150.
- Minetoki T, Gomi K, Kitamoto K, et al. Nucleotide sequence and expression of α-glucosidase-encoding gene (agdA) from Aspergillus oryzae. Biosci Biotechnol Biochem. 1995;59:1516–1521.
- Tada S, Kitamoto K, Gomi K, et al. Identification of the promoter region of the Taka-amylase A gene required for starch induction. Agric Biol Chem. 1991;55:1939–1941.
- Tsuchiya K, Tada S, Gomi K, et al. Deletion analysis of the Taka-amylase A gene promoter using a homologous transformation system in Aspergillus oryzae. Biosci Biotechnol Biochem. 1992;56:1849–1853.
- Kanemori Y, Gomi K, Kitamoto K, et al. Insertion analysis of putative functional elements in the promoter region of the Aspergillus oryzae Taka-amylase A gene (amyB) using a heterologous Aspergillus nidulans amdS-lacZ fusion gene system. Biosci Biotechnol Biochem. 1999;63:180–183.
- Hata Y, Kitamoto K, Gomi K, et al. Functional elements of the promoter region of the Aspergillus oryzae glaA gene encoding glucoamylase. Curr Genet. 1992;22:85−91.
- Minetoki T, Nunokawa Y, Gomi K, et al. Deletion analysis of promoter elements of the Aspergillus oryzae agdA gene encoding a-glucosidases. Curr Genet. 1996;30:432−438.
- Minetoki T, Kumagai C, Gomi K, et al. Improvement of promoter activity by the introduction of multiple copies of the conserved region III sequence, involved in the efficient expression of Aspergillus oryzae amylase-encoding genes. Appl Microbiol Biotechnol. 1998;50:459–467.
- Gomi K, Akeno T, Minetoki T, et al. Molecular cloning and characterization of a transcriptional activator gene, amyR, involved in the amylolytic gene expression in Aspergillus oryzae. Biosci Biotechnol Biochem. 2000;64:816–827.
- Machida M, Asai K, Sano M, et al. Genome sequencing and analysis of Aspergillus oryzae. Nature. 2005;438:1157–1161.
- Mizutani O, Kudo Y, Saito A, et al. A defect of LigD (human Lig4 homolog) for nonhomologous end joining significantly improves efficiency of gene-targeting in Aspergillus oryzae. Fungal Genet Biol. 2008;45:878–889.
- Punt PJ, Schuren FH, Lehmbeck J, et al. Characterization of the Aspergillus niger prtT, a unique regulator of extracellular protease encoding genes. Fungal Genet Biol. 2008;45:1591–1599.
- Sharon H, Hagag S, Osherov N. Transcription factor PrtT controls expression of multiple secreted proteases in the human pathogenic mold Aspergillus fumigatus. Infect Immun. 2009;77:4051–4060.
- Needleman RB, Kaback DB, Dubin RA, et al. MAL6 of Saccharomyces: a complex genetic locus containing three genes required for maltose fermentation. Proc Natl Acad Sci USA. 1984;81:2811–2815.
- Needleman RB. Control of maltase synthesis in yeast. Mol Microbiol. 1991;5:2079–2084.
- Sirenko OI, Ni B, Needleman RB. Purification and binding properties of the Mal63p activator of Saccharomyces cerevisiae. Curr Genet. 1995;27:509–516.
- Petersen KL, Lehmbeck J, Christensen J. A new transcriptional activator for amylase genes in Aspergillus. Mol Gen Genet. 1999;262:668–676.
- Tani S, Itoh T, Kato M, et al. In vivo and in vitro analyses of the AmyR binding site of the Aspergillus nidulans agdA promoter; requirement of the CGG direct repeat for induction and high affinity binding of AmyR. Biosci Biotechnol Biochem. 2001;65:1568–1574.
- Ito T, Tani S, Itoh T, et al. Mode of AmyR binding to the CGGN8AGG sequence in the Aspergillus oryzae taaG2 promoter. Biosci Biotechnol Biochem. 2004;68:1906–1911.
- Wang P, Kojima T, Kobayashi T, et al. Comprehensive analysis of the DNA-binding specificity of an Aspergillus nidulans transcription factor, AmyR, using a bead display system. Biosci Biotechnol Biochem. 2012;76:1128–1134.
- Tani S, Katsuyama Y, Hayashi T, et al. Characterization of the amyR gene encoding a transcriptional activator for the amylase genes in Aspergillus nidulans. Curr Genet. 2001;39:10–15.
- Akao T, Sano M, Yamada O, et al. Analysis of expressed sequence tags from the fungus Aspergillus oryzae cultured uder different conditions. DNA Res. 2007;14:47–57.
- Hasegawa S, Takizawa M, Suyama H, et al. Characterization and expression analysis of a maltose-utilizing (MAL) cluster in Aspergillus oryzae. Fungal Genet Biol. 2010;47:1–9.
- Medintz I, Wang X, Hradek T, et al. A PEST-like sequence in the N-terminal cytoplasmic domain of Saccharomyces maltose permease is required for glucose-induced proteolysis and rapid inactivation of transport activity. Biochemistry. 2000;39:4518–4526.
- Hiramoto T, Tanaka M, Ichikawa T, et al. Endocytosis of a maltose permease is induced when amylolytic enzyme production is repressed in Aspergillus oryzae. Fungal Genet Biol. 2015;82:136–144.
- Kato N, Murakoshi Y, Kato M, et al. Isomaltose formed by α-glucosidases triggers amylase induction in Aspergillus nidulans. Curr Genet. 2002;42:43–50.
- Kato N, Suyama S, Shirokane M, et al. Novel α-glucosidase from Aspergillus nidulans with strong transglycosylation activity. Appl Environ Microbiol. 2002;68:1250–1256.
- Makita T, Katsuyama Y, Tani S, et al. Inducer-dependent nuclear localization of a Zn(II)2Cys6 transcriptional activator, AmyR, in Aspergillus nidulans. Biosci Biotechnol Biochem. 2009;73:391–399.
- Murakoshi Y, Makita T, Kato M, et al. Comparison and characterization of α-amylase inducers in Aspergillus nidulans based on nuclear localization of AmyR. Appl Microbiol Biotechnol. 2012;94:1629–1635.
- Suzuki K, Tanaka M, Konno Y, et al. Distinct mechanism of activation of two transcription factors, AmyR and MalR, involved in amylolytic enzyme production in Aspergillus oryzae. Appl Microbiol Biotechnol. 2015;99:1805–1815.
- Vongsangnak W, Salazar M, Hansen K, et al. Genome-wide analysis of maltose utilization and regulation in aspergilli. Microbiology. 2009;155:3893–3902.
- Nakamura T, Maeda Y, Tanoue N, et al. Expression profile of amylolytic genes in Aspergillus nidulans. Biosci Biotechnol Biochem. 2006;70:2363–2370.
- Bali M, Zhang B, Morano KA, et al. The Hsp90 molecular chaperone complex regulates maltose induction and stability of the Saccharomyces MAL gene transcription activator Mal63p. J Biol Chem. 2003;278:47441–47448.
- Ran F, Bali M, Michels CA. Hsp90/Hsp70 chaperone machine regulation of the Saccharomyces MAL-activator as determined in vivo using non inducible and constitutive mutant alleles. Genetics. 2008;179:331–343.
- Konno Y, Suzuki K, Tanaka M, et al. Chaperone complex formation of the transcription factor MalR involved in maltose utilization and amylolytic enzyme production in Aspergillus oryzae. Biosci Biotechnol Biochem. 2018;82:827–835.
- Wandinger SK, Richter K, Buchner J. The Hsp90 chaperone machinery. J Biol Chem. 2008;283:18473–18477.
- Li J, Soroka J, Buchner J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biophys Biochim Acta. 2012;1823:624–635.
- Hata Y, Ishida H, Kojima Y, et al. Comparison of two glucoamylase produced by Aspergillus oryzae in solid-state culture (koji) and in submerged culture. J Ferment Bioeng. 1997;84:532–537.
- Hata Y, Ishida H, Ichikawa E, et al. Nucleotide sequence of an alternative glucoamylase-encoding gene (glaB) expressed in solid-state culture of Aspergillus oryzae. Gene. 1998;207:127–134.
- Gomi K, Arikawa K, Kamiya N, et al. Cloning and nucleotide sequence of the acid protease-encoding gene (pepA) from Aspergillus oryzae. Biosci Biotechnol Biochem. 1993;57:1095–1100.
- Kitano H, Kataoka K, Furukawa K, et al. Specific expression and temperature-dependent expression of the acid protease-encoding gene (pepA) in Aspergillus oryzae in solid-state culture (Rice-Koji). J Biosci Bioeng. 2002;93:563–567.
- Obata H, Ishida H, Hata Y, et al. Cloning of a novel tyrosinase-encoding gene (melB) from Aspergillus oryzae and its overexpression in solid-state culture (Rice Koji). J Biosci Bioeng. 2004;97:400–405.
- Watanabe J, Tanaka H, Mogi Y, et al. Loss of Aspergillus oryzae amyR function indirectly affects hemicellulolytic and cellulolytic enzyme production. J Biosci Bioeng. 2011;111:408–413.
- Jin FJ, Nishida M, Hara S, et al. Identification and characterization of a putative basic helix-loop-helix transcription factor involved in the early stage of conidiophore development in Aspergillus oryzae. Fungal Genet Biol. 2011;48:1108–1115.
- Ogawa M, Kobayashi T, Koyama Y. ManR, a novel Zn(II)2Cys6 transcriptional activator, controls the β-mannan utilization system in Aspergillus oryzae. Fungal Genet Biol. 2012;49:987–995.
- Ishida H, Hata Y, Ichikawa E, et al. Regulation of the glucoamylase-encoding gene (glaB), expressed in solid-state culture (koji) of Aspergillus oryzae. J Ferment Bioeng. 1998;86:301–307.
- Tanaka M, Yoshimura M, Ogawa M, et al. The C2H2-type transcription factor, FlbC, is involved in the transcriptional regulation of Aspergillus oryzae glucoamylase and protease genes specifically expressed in solid-state culture. Appl Microbiol Biotechnol. 2016;100:5859–5868.
- Kwon NJ, Garzia A, Espeso EA, et al. FlbC is a putative nuclear C2H2 transcription factor regulating development in Aspergillus nidulans. Mol Microbiol. 2010;77:1203–1219.
- Ogawa M, Tokuoka M, Jin FJ, et al. Genetic analysis of conidiation regulatory pathways in koji-mold Aspergillus oryzae. Fungal Genet Biol. 2010;47:10–18.
- Park HS, Yu JH. Genetic control of asexual sporulation in filamentous fungi. Curr Opin Microbiol. 2012;15:669–677.
- Hisada H, Sano M, Ishida H, et al. Identification of regulatory elements in the glucoamylase-encoding gene (glaB) promoter from Aspergillus oryzae. Appl Microbiol Biotechnol. 2013;97:4951–4956.
- Ishida H, Hata Y, Kawato A, et al. Identification of functional elements that regulate the glucoamylase-encoding gene (glaB) expressed in solid-state culture of Aspergillus oryzae. Curr Genet. 2000;37:373–379.
- Boni AC, Ambrósio DL, Cupertino FB, et al. Neurospora crassa developmental control mediated by the FLB-3 transcription factor. Fungal Biol. 2018;122:570–582.
- Matheis S, Yemelin A, Scheps D, et al. Functions of the Magnaporthe oryzae Flb3p and Flb4p transcription factors in the regulation of conidiation. Microbiol Res. 2017;196:106–117.
- Dowzer CE, Kelly JM. Cloning of the creA gene from Aspergillus nidulans: a gene involved in carbon catabolite repression. Curr Genet. 1989;15:457–459.
- Lockington RA, Kelly JM. Carbon catabolite repression in Aspergillus nidulans involves deubiquitination. Mol Microbiol. 2001;40:1311–1321.
- Lockington RA, Kelly JM. The WD40-repeat protein CreC interacts with and stabilizes the deubiquitinating enzyme CreB in vivo in Aspergillus nidulans. Mol Microbiol. 2002;43:1173–1182.
- Boase N, Kelly JM. A role for creD, a carbon catabolite repression gene from Aspergillus nidulans, in ubiquitination. Mol Microbiol. 2004;53:929–940.
- Ichinose S, Tanaka M, Shintani T, et al. Improved α-amylase production by Aspergillus oryzae after a double deletion of genes involved in carbon catabolite repression. Appl Microbiol Biotechnol. 2014;98:335–343.
- Nakari-Setälä T, Paloheimo M, Kallio J, et al. Genetic modification of carbon catabolite repression in Trichoderma reesei for improved protein production. Appl Environ Microbiol. 2009;75:4853–4860.
- Ichinose S, Tanaka M, Shintani T, et al. Increased production of biomass-degrading enzymes by double deletion of creA and creB genes involved in carbon catabolite repression in Aspergillus oryzae. J Biosci Bioeng. 2018;125:141–147.
- Noguchi Y, Sano M, Kanamaru K, et al. Genes regulated by AoXlnR, the xylanolytic and cellulolytic transcriptional regulator, in Aspergillus oryzae. Appl Microbiol Biotechnol. 2009;85:141–154.
- de Assis LJ, Ries LN, Savoldi M, et al. Aspergillus nidulans protein kinase A plays an important role in cellulase production. Biotechnol Biofuels. 2015;8:213.
- Kunitake E, Li Y, Uchida R, et al. CreA-independent carbon catabolite repression of cellulase genes by trimeric G-protein and protein kinase A in Aspergillus nidulans. Curr Genet. 2019. DOI:10.1007/s00294-019-00944-4.
- Miyazawa K, Yoshimi A, Zhang S, et al. Increased enzyme production under liquid culture conditions in the industrial fungus Aspergillus oryzae by disruption of the genes encoding cell wall α-1,3-glucan synthase. Biosci Biotechnol Biochem. 2016;80:1853–1863.
- Zhang S, Sato H, Tanaka M, et al. Cell wall α-1,3-glucan prevents α-amylase adsorption onto the fungal cell in submerged culture in Aspergillus oryzae. J Biosci Bioeng. 2017;125:141–147.
- Yoshimi A, Sano M, Inaba A, et al. Functional analysis of the α-1,3-glucan synthase genes agsA and agsB in Aspergillus nidulans: agsB is the major α-1,3-glucan synthase in this fungus. PLoS One. 2013;8:e54893.
- Fontaine T, Beauvais A, Loussert C, et al. Cell wall α-1-3glucans induce the aggregation of germinating conidia of Aspergillus fumigatus. Fungal Genet Biol. 2010;47:707–712.
- He X, Li S, Kaminskyj SGW. Characterization of Aspergillus nidulans α-glucan synthesis: roles for two synthases and two amylases. Mol Microbiol. 2014;91:579–595.
- He X, Li S, Kaminskyj SGW. Overexpression of Aspergillus nidulans α-1,3-glucan synthase increases cellular adhesion and causes cell wall defects. Med Mycol. 2018;56:645–648.
- Miyazawa K, Yoshimi A, Kasahara S, et al. Molecular mass and localization of α-1,3-glucan in cell wall control the degree of hyphal aggregation in liquid culture of Aspergillus nidulans. Front Microbiol. 2018;9:2623.
- Thevelein J, Voordeckers K. Functioning and evolutionary significance of nutrient transceptors. Mol Biol Evol. 2009;26:2407–2414.
- Van Dijck P, Brown NA, Goldman GH, et al. Nutrient sensing at the plasma membrane of fungal cells. Microbiol Spectr. 2017;5:FUNK-0031–2016.
- Znameroski EA, Li X, Tsai JC, et al. Evidence for transceptor function of cellodextrin transporters in Neurospora crassa. J Biol Chem. 2014;289:2610–2619.
- Zhang W, Kou Y, Xu J, et al. Two major facilitator superfamily sugar transporters from Trichoderma reesei and their roles in induction of cellulose biosynthesis. J Biol Chem. 2013;288:32861–32872.
- Cai P, Wang B, Ji J, et al. The putative cellodextrin transporter-like protein CLP1 is involved in cellulase induction in Neurospora crassa. J Biol Chem. 2015;290:788–796.
- Hong SB, Lee M, Kim DH, et al. Aspergillu luchuensis, an industrially important black Aspergillus in east Asia. PLoS One. 2013;8:e63769.
- Hong SB, Yamada O, Samson RA. Taxinomic re-evaluation of black koji mold. Appl Microbiol Biotechnol. 2014;98:555–561.
- Pel HJ, de Winde JH, Archer DB, et al. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol. 2007;25:221–231.
- Yamada O, Machida M, Hosoyama A, et al. Genome sequence of Aspergillus luchuensis NBRC 4314. DNA Res. 2016;23:507–515.
- Yuan XL, van der Kaaij RM, van den Hondel C, et al. Aspergillus niger genome-wide analysis reveals a large number of novel α-glucan acting enzymes with unexpected expression profiles. Mol Genet Genomics. 2008;279:545–561.
- Nagamine K, Murashima K, Kato T, et al. Mode of α-amylase production by the shochu koji mold Aspergillus kawachii. Biosci Biotechnol Biochem. 2003;67:2194–2202.
- Hagiwara D, Sakamoto K, Abe K, et al. Signaling pathways for stress responses and adaptation in Aspergillus species: stress biology in the post-genomic era. Biosci Biotechnol Biochem. 2016;80:1667–1680.
- Tanaka M, Hiramoto T, Tada H, et al. Improved α-amylase production by dephosphorylation mutation of CreD, an arrestin-like protein required for glucose-induced endocytosis of maltose permease and carbon catabolite de-repression in Aspergillus oryzae. Appl Environ Microbiol. 2017;83:e00592–17.
- Ries LN, Beattie SR, Espeso EA, et al. Diverse regulation of the CreA carbon catabolite repressor in Aspergillus nidulans. Genetics. 2016;203:335–352.
- Tanaka M, Ichinose S, Shintani T, et al. Nuclear export-dependent degradation of the carbon catabolite repressor CreA is regulated by a region located near the C-terminus in Aspergillus oryzae. Mol Microbiol. 2018;110:176–190.
- Alam MA, Kamlangdee N, Kelly JM. The CreB deubiquitinating enzyme does not directly target the CreA repressor protein in Aspergillus nidulans. Curr Genet. 2017;63:647–667.
- Alam MA, Kelly JM. Proteins interacting with CreA and CreB in the carbon catabolite repression network in Aspergillus nidulans. Curr Genet. 2017;63:669–683.