
ABSTRACT
This study aims to study the effects of adenosine A2A receptor (A2AR) on hippocampal cell apoptosis and the putative mechanisms in a mouse model of chronic hypoxic-hypercapnia. Wild-type (WT) or A2AR knockout (A2AR KO) mice were randomly divided into normal control (NC) groups and chronic hypoxic-hypercapnia (4HH) groups. Compared with their corresponding NC groups (WT-NC and KO-NC), the apoptosis index (AI), caspase-3 activity, Bax mRNA and P-p38 protein expression in the hippocampus of 4HH groups (WT-4HH and KO-4HH) were significantly increased, while Bcl2 mRNA expression was significantly decreased (P < 0.05). Moreover, A2AR deficiency significantly rescued the effect of chronic hypoxic-hypercapnia on apoptosis when compared with the WT-4HH group (P < 0.05). A2AR deficiency inhibits hippocampal cell apoptosis in mice exposed to chronic hypoxic-hypercapnia, which might be associated with dampened p38 MAPK activation and Bax mRNA expression, and augmented Bcl-2 mRNA expression.
Graphical abstract
TUNEL staining in the hippocampus CA1 area of each group.
Chronic obstructive pulmonary disease (COPD) is an important globally-prevalent cause of morbidity and mortality [Citation1,Citation2]. Recently, cognitive impairment is recognized as an important extra-pulmonary manifestation in patients with COPD [Citation3,Citation4]. Chronic hypoxic-hypercapnia is a common pathophysiological phenomenon in COPD [Citation5]. Furthermore, blood carbon dioxide levels predict cognitive deficits in COPD patients, and hypoxia in COPD results in a relatively focused pattern of impaired measurement of memory function and tasks requiring attention allocation [Citation6,Citation7].
The chronic hypoxic-hypercapnia model can mimic the pathophysiology of COPD [Citation8]. Lin et al. observed that exposure to hypoxic-hypercapnia impaired cognition in a rat model [Citation9]. Further, Shao et al. found that neuronal apoptosis in the hippocampus might play an important role in the chronic hypoxic-hypercapnia model [Citation10].
Adenosine is an important transmitter of the central nervous system, which mainly acts through two metabotropic adenosine receptors with opposite functions, namely A1 and A2A receptors (A1Rs, and A2ARs) [Citation11]. Increased workload and decreased metabolic ability of brain tissues can markedly increase the levels of adenosine [Citation12]. Adenosine has dual effects in the adult brain [Citation13]. The initial activation of A1Rs is essentially required for brain damage; however, over-activation of A1Rs leads to the desensitization of A1Rs [Citation4]; thus, A2ARs contribute to aggravated brain damage in adults [Citation14]. In vitro studies showed that blockade of adenosine A2ARs prevented staurosporine-induced apoptosis of hippocampal neurons in the brain and synaptic toxicity caused by β-amyloid peptides [Citation15,Citation16]. Li et al. found that genetic inactivation of the adenosine A2AR prevented apoptosis in the mouse cortical impact model [Citation17]. Therefore, A2ARs might directly regulate neuronal apoptosis, even though the underlying mechanism remains unclear.
It is reported that A2ARs expressed in hippocampal microglia was involved in microglia activation and neuroinflammation, which further mediated spatial memory impairment under acute hypobaric hypoxia [Citation18]. In chronic neurodegenerative conditions, the blockade of A2ARs affords protection against Parkinson’s, Alzheimer’s, or Huntington’s disease [Citation19–Citation21], which all involve features of apoptotic neurons [Citation22]. This neuroprotection by A2AR inactivation is largely attributed to inhibition of excitatory neurotransmitter release, inflammatory cytokine production and oxygen free radical release [Citation23,Citation24]. However, the role of A2AR in chronic hypoxic-hypercapnia has not been studied.
In this study, we investigated the effects of adenosine A2AR on the apoptosis of hippocampal cells in a mouse model of chronic hypoxic-hypercapnia and the underlying mechanisms.
Materials and methods
Animals
C57BL/6 mice used in this study were originally from Dr. Jiangfan Chen’s laboratory (Boston University of School Medicine, MA, USA) and bred by the Experimental Animal Center of Wenzhou Medical University (Wenzhou, China). The A2AR+/− breeding pairs were backcrossed to C57BL/6 mice for 10 generations, resulting in a congenic C57BL/6 genetic background. The generation of the KO mice and their genotyping was conducted as described previously [Citation25]. Compared to WT mice under basal conditions, no gross anatomical or histological abnormality and no significant changes in relevant neuro-chemical systems or physiologic parameters were found in the initial characterization of the A2AR KO mice [Citation26]. The mice were housed under 12 h light/dark conditions with free access to food and water. Male mice at 9–10 months of age were used in this study. The procedure for this experiment was approved by the Animal Care Committee of Wenzhou Medical University.
Experimental groups and treatment
Sixteen male WT mice and sixteen A2AR KO mice were randomly divided into two groups, respectively, with 8 mice per group: the hypoxic-hypercapnia groups (WT-4HH, and KO-4HH) and the normal control groups (WT-NC, and KO-NC). The WT-4HH, and KO-4HH groups were kept in a sealed and ventilated chamber containing 9% – 11% O2 and 5% – 6% CO2 8 hours a day, 6 days a week, for 4 weeks [Citation27]. During other time, mice in all groups were housed in the same room. After 4 weeks, all animals were anesthetized by intra-peritoneal sodium pentobarbital (100 mg/kg), perfused from the heart and then sacrificed by decapitation. Both sides of the hippocampus were then rapidly separated. The left hippocampus were stored in liquid nitrogen for following qRT-PCR, Caspase-3 activity and Western blot analyses while the right hippocampus were fixed in 4% paraformaldehyde for TUNEL assay.
TUNEL assay
TUNEL was performed using the TUNEL-pod kit (Beyotime Institute of Biotechnology, Shanghai, China). Briefly, the hippocampus sections were incubated with 3% H2O2 for inactivation of endogenous hydrogen peroxidase activity. After rinsing with PBS, the sections were incubated with proteinase K solution at 37°C for 20 min to enhance the permeability. Then, the sections were incubated for 60 min with TUNEL reaction mixture and for 30 min with converter-POD at 37°C. At last, after incubation for 10 min with DAB substrate solution (Zymed, CA, USA), the sections were counterstained with 0.5% methyl green. The FITC-labeled TUNEL-positive cells (green) were observed and calculated under CX31 fluorescent microscopy (OLYMPUS, Tokyo, Japan). The hippocampus CA1 region was found under the microscope. Positive and negative controls were carried out on slides from the same block. Stained slides were randomly observed under a high magnification microscope. The image pictures were processed by the America IPP6.0 software and the positive cells (green) were calculated as apoptosis index (AI).
Protein extraction
The frozen hippocampus samples were ground and lysed in RIPA lysis buffer containing a protease inhibitor cocktail (Roche) and PMSF. Protein concentrations were determined using the Bio-Rad Protein Assay reagent procedure (Bio-Rad, Hercules, CA, USA).
Caspase-3 activity assay
Caspase-3 activity was measured by cleavage of chromogenic caspase substrates, Ac-DEVD-pNA (acetyl-Asp-Glu-Val-Asp p-nitroanilide) and Ac-LEHD-pNA (acetyl-Leu-Glu-His-Asp p-nitroanilide), and by using the Caspase-3 kit (Beyotime Institute of Biotechnology, Shanghai, China). Approximately 50 μg of total protein was added to reaction buffer containing Ac-DEVD-pNA (2mM) or Ac-LEHD-pNA (2mM), and incubated for 2h at 37°C. The absorbance of yellow pNA cleavage from its corresponding precursors was measured by a spectrometer (UVP, CA, USA) at a wavelength of 405 nm.
Quantitative PCR
Total RNA from the hippocampus was extracted using Trizol reagent (Thermo Fisher, MA, USA), and cDNA was synthesized using the first-strand cDNA synthesis kit (Thermo Fisher). PCR was performed with a Light-Cycler 480 thermal cycler (Roche, USA) using SYBR® Premix Ex TaqTM II (TaKaRa, Dalian, China). The sequences of primers were designed and synthesized by TaKaRa ().
Table 1. The sequence of the primers used for qPCR.
The PCR procedure was as follows: Pre-incubation: 95°C, 30S for 1 cycle; PCR: 95°C, 5S, 56°C 30S, 72°C, 30S, for 40 cycles; Melting: 95°C, 1S, 65°C, 15S, 1 Cycle; Cooling: 40°C, 30S, 1 Cycle. The relative concentration of each gene for each sample was estimated by the Light Cycler 480 software (Roche) using the 2−ΔΔdT method with GAPDH as the internal control.
Western blotting analysis
Samples (50 μg of protein per well) were loaded onto a 10% SDS-PAGE gel and resolved by standard electrophoresis. The proteins were then transferred electrophoretically onto nitrocellulose membrane (Hybond-C extra; Amersham, Arlington Heights, IL, USA). Membranes were blocked for 1h at room temperature with 5% non-fat milk in PBS and then probed for 2 h at room temperature using a primary antibody against p38MAPK (rabbit polyclonal, 1:2000; New England Biolabs (NEB), Beverly, MA, USA), phospho-p38MAPK (rabbit polyclonal, 1:2000; NEB), or beta- tubulin (1:5000; NEB). The membranes were then incubated with horseradish peroxidase-conjugated anti-rabbit IgG (1:5000; Boehringer Mannheim, Indianapolis, IN, USA). The bands were visualized using a chemilumine scence-based detection kit (Pierce Company, USA). The OD value was quantified using Quantity One gel analysis system (Amersham ECL Western Blotting Analysis System, GE Healthcare, USA) and quantitated by densitometry.
Statistical analysis
All data was processed using SPSS16.0 statistics software. All values were expressed as mean ±standard deviation. Kolmogorov-Smirnov test was performed to all data. The multi-group data were analyzed using the signal factor analysis of variance (one-way ANOVA). Pair wise comparison was performed using LSD test for homogeneous variance, and Dunnett’s T3 test for nonhomogeneous variance. In all cases, P < 0.05 was considered significantly.
Results
A2AR deficiency decreases the apoptosis of hippocampus cells
We first analyzed whether A2AR can regulate hippocampus cell apoptosis. As determined by TUNEL assay, the AI of the hippocampus was significantly increased in both 4HH groups as compared with their corresponding control groups (P < 0.05). In addition, more apoptotic cells were found in the WT-4HH group than in the KO-4HH group (P < 0.05) (), indicating the essential role of A2AR in promoting cell apoptosis. Caspase-3 is known as the final executor of cellular apoptosis, and caspase-3 activation is a prime indicator of apoptosis [Citation28]. We also found that the hippocampal caspase-3 activity in the two 4HH groups was significantly higher than that in corresponding NC groups (P < 0.05). Importantly, caspase-3 activity in the WT-4HH group was significantly higher than that in the KO-4HH group (P < 0.05) (). These results suggest that A2AR deficiency decreases the apoptosis of hippocampus cells.
Figure 1. TUNEL staining in the hippocampus CA1 area of each group.
(a) Representative photographs of TUNEL staining in the hippocampus CA1 area of each group (Scale bar = 25 μM). (b) Apoptosis index (AI) of hippocampus in various groups as shown in (a). #p < 0.01 vs KO-NC group; *p < 0.01 vs WT-NC group; △p < 0.05 vs KO-4HH group.
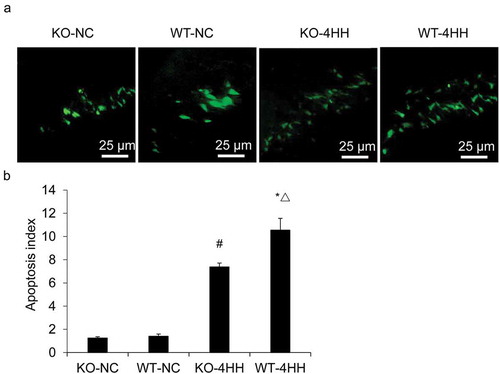
Figure 2. Apoptosis analysis of mouse hippocampus cells.
Caspase-3 activity of hippocampus in various groups. #p < 0.01 vs KO-NC group; *p < 0.01 vs WT-NC group; △p < 0.05 vs KO-4HH group.
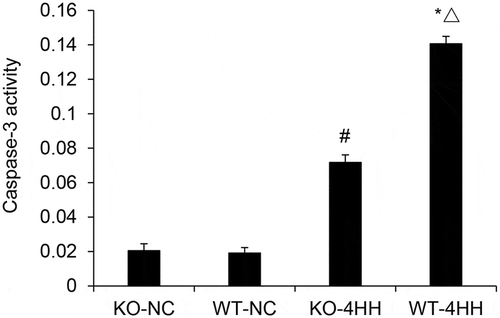
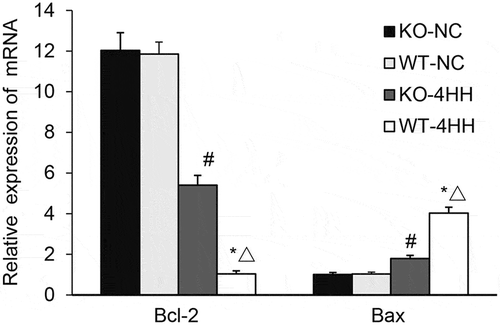
Effect of A2AR on the expression of Bcl-2 and Bax
The expressions of Bcl-2 mRNA and Bax mRNA were detected with quantitative PCR. The results showed that hypoxic-hypercapnia treatment significantly decreased the expression of Bcl-2 mRNA while increased the expression of Bax mRNA () in the hippocampus as compared with their corresponding control groups (P < 0.05). Further, we found that in the 4HH groups, A2AR KO dramatically reduced Bax expression but increased Bcl-2 expression when compared with the WT mice (P < 0.05). These results suggest that A2AR deficiency inhibited Bax mRNA expression and augmented Bcl-2 mRNA expression.
Figure 3. qPCR Analysis of Bcl-2 and Bax mRNA expression in mouse hippocampus tissues.
Expression of Bcl-2 mRNA and Bax mRNA in mouse hippocampus tissues of various groups. #p < 0.01 vs KO-NC group; *p < 0.01 vs WT-NC group; △p < 0.05 vs KO-4HH group.
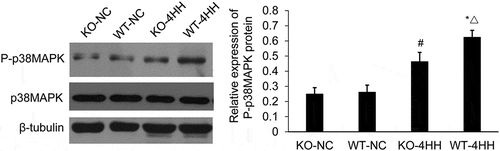
A2AR deficiency increases the expression of p-p38MAPK protein
Finally, we analyzed p38MAPK and p-p38MAPK expression by western blot to further investigate the possible mechanisms for the regulation of A2AR on apoptosis. The level of p38MAPK protein was not obviously changed among groups (). However, the protein expression of p-p38 MAPK in the hippocampus was higher after hypoxic-hypercapnia treatment. Moreover, this elevation was partially impaired by A2AR KO, showing lower expression of p-p38 MAPK when compared with the WT-4HH group in the hippocampus (P < 0.05) (). These results suggest that A2AR deficiency dampens the expression of p-p38MAPK protein.
Figure 4. Western blot analysis of the expression of p38MAPK and P-p38MAPK protein in hippocampus of various groups.
#p < 0.01 vs KO-NC group; *p < 0.01 vs WT-NC group; △p < 0.05 vs KO-4HH group.
Discussion
In the present investigation, apoptotic cells could be detected in both normal and chronic hypoxic-hypercapnia mice by TUNEL staining, an observation that is consistent with the findings by Shao et al. [Citation10]. More importantly, we found that A2AR KO can inhibit hippocampal apoptosis in the murine chronic hypoxic-hypercapnia model.
We also demonstrated that deletion of A2AR enhanced the mRNA expression of the anti-apoptotic protein Bcl-2 and decreased the mRNA expression of the pro-apoptotic protein Bax, thus resulting in reduced caspase activation. Mice in the chronic hypoxic-hypercapnia model groups had increased hippocampal caspase-3 activity. The interaction between the anti-apoptotic protein Bcl-2 and the pro-apoptotic protein Bax play key roles in activating intrinsic apoptotic signals that involve release of cytochrome C by mitochondria and activation of intracellular cysteine proteases, particularly caspase-9 and caspase-3 [Citation29,Citation30]. Silva et al. [Citation31] noted that blockade of A2ARs considerably reduced Staurosporine (STS)-induced membrane leakage and prevented the release of cytochrome C from mitochondria.
MAPKs (mitogen-activated protein kinases) are a class of serine threonine protein kinases that are widely distributed in mammalian cells and play an essential role in extracellular signal transduction [Citation32]. Thus far, four types of MAPKs have been identified: 1) Extracellular signal-regulated kinases (ERK1/2); 2) c-Jun N-terminal kinase (JNK)/Stress-activated protein kinase (SAPK); 3) p38 MAPK and 4) ERK5 [Citation32]. Indeed, there are extensive interactions between the MAPK signal pathways and the adenosine receptors, and in turn, all of the adenosine receptors may activate at least one subfamily of the MAPKs [Citation33]. p38 MAPK (p38) is a stress-activated protein kinase that can induce apoptosis under stress conditions [Citation34], and the inhibition of p38 can suppress apoptosis provoked various stimuli [Citation35].
P38 MAPK can activate Bax, which further changes the permeability of mitochondrial membrane and eventually causes apoptosis [Citation36]. Therefore we speculate that the decreased expression of hippocampal Bax mRNA in the chronic hypoxic-hypercapnia model of A2AR KO mice may be related to the inhibition of p38 MAPK signal transduction pathway. Indeed, A2AR deficiency decreased the level of phosphorylated p38 MAPK expression, but not p38MPK expression, in the hippocampus of chronic hypoxic-hypercapnia mice. It is reported that specific adenosine A2AR antagonist can inactivate neuronal p38 MAPK, and inhibit ischemia-reperfusion and the expression of the Aβ protein [Citation16,Citation37]. Therefore, our findings might represent a mechanism depending on decreased expression of phosphorylated p38, altered anti-apoptotic Bcl-2 and pro-apoptotic Bax expression that is involved in hippocampal apoptosis in the hypoxic-hypercapnia condition.
In conclusion, the present study shows that adenosine A2AR KO exhibits significant anti-apoptotic effects in the hippocampus cells of mice exposed to chronic hypoxic hypercapnia condition. This effect is associated with decreased phosphorylation of p38 and altered anti-apoptotic Bcl-2 and pro-apoptotic Bax expression. This observation suggests that adenosine A2AR antagonists might have therapeutic value in reducing damage to brain function induced by chronic hypoxic-hypercapnia.
Author contributions
XW designed the study, searched the literatures and prepared the manuscript. HR conducted the experiments, performed the statistical analysis and prepared the manuscript. ZC conducted the experiments and performed the statistical analysis. XG collected the funds. All authors have read and approved the final manuscript.
Data availability
The data that support the findings of this study are available on request from the corresponding author.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Rosenberg SR, Kalhan R, Mannino DM. Epidemiology of chronic obstructive pulmonary disease: prevalence, morbidity, mortality, and risk factors. Semin Respir Crit Care Med. 2015 Aug;36(4):457–469.
- Shield KD, Rehm J. Global risk factor rankings: the importance of age-based health loss inequities caused by alcohol and other risk factors. BMC Res Notes. 2015 Jun;9(8):231.
- Cleutjens FA, Janssen DJ, Ponds RW, et al. COgnitive-pulmonary disease. Biomed Res Int. 2014;2014:697825.
- Singh B, Parsaik AK, Mielke MM, et al. Chronic obstructive pulmonary disease and association with mild cognitive impairment: the Mayo Clinic Study of Aging. Mayo Clin Proc. 2013 Nov;88(11):1222–1230.
- Min JJ, Huo XL, Xiang LY, et al. Protective effect of Dl-3n-butylphthalide on learning and memory impairment induced by chronic intermittent hypoxia-hypercapnia exposure. Sci Rep. 2014 Jul 3;4:5555.
- Klein M, Gauggel S, Sachs G, et al. Impact of chronic obstructive pulmonary disease (COPD) on attention functions. Respir Med. 2010 Jan;104(1):52–60.
- Stuss DT, Peterkin I, Guzman DA, et al. Chronic obstructive pulmonary disease: effects of hypoxia on neurological and neuropsychological measures. J Clin Exp Neuropsychol. 1997 Aug;19(4):515–524.
- Zheng GQ, Wang Y, Wang XT. Chronic hypoxia-hypercapnia influences cognitive function: a possible new model of cognitive dysfunction in chronic obstructive pulmonary disease. Med Hypotheses. 2008;71(1):111–113.
- Lin YY, Chen KY, Huo XL, et al. Cholinergic dysfunctions are correlated with memory deficits in rats exposed to chronic hypoxia-hypercapnia. Chinese J of Neurology. 2013;46:47–50.
- Shao SM, Chen SF, Yong LI, et al. Effect of chronic hypoxic hypercapnia on the expression of Bcl-2 and Bax genes and apoptosis in rat hippocampal cells. J Wenzhou Med Col. 2008:1:12–14.
- Venton B, Nguyen ME, Ross A. Mechanism and function of transient adenosine release in the brain. Purinergic Signalling. 2014;10:661–664.
- Dulla CG, Masino SA. Physiologic and metabolic regulation of adenosine: mechanisms. New York: Springer New York; 2013. ( (S. Masino, Boison D, editors. Adenosine: A Key Link between Metabolism and Brain Activity)).
- Saransaari P, Oja SS. Involvement of nitric oxide in adenosine release in the developing and adult mouse hippocampus. Neurochem Res. 2004 Jan;29(1):219–225.
- Dos Santos-Rodrigues A, Pereira MR, Brito R, et al. Adenosine transporters and receptors: key elements for retinal function and neuroprotection. Vitam Horm. 2015;98:487–523.
- Haller S, Montandon ML, Rodriguez C, et al. Acute caffeine administration effect on brain activation patterns in mild cognitive impairment. J Alzheimers Dis. 2014;41(1):101–112.
- Simoes AP, Duarte JA, Agasse F, et al. Blockade of adenosine A2A receptors prevents interleukin-1beta-induced exacerbation of neuronal toxicity through a p38 mitogen-activated protein kinase pathway. J Neuroinflammation. 2012 Aug 20;9:204.
- Li W, Dai S, An J, et al. Genetic inactivation of adenosine A2A receptors attenuates acute traumatic brain injury in the mouse cortical impact model. Exp Neurol. 2009 Jan;215(1):69–76.
- Chen PZ, He WJ, Zhu ZR, et al. Adenosine A2A receptor involves in neuroinflammation-mediated cognitive decline through activating microglia under acute hypobaric hypoxia. Behav Brain Res. 2018 Jul 16;347:99–107.
- Chen JF, Chern Y. Impacts of methylxanthines and adenosine receptors on neurodegeneration: human and experimental studies. Handb Exp Pharmacol. 2011;200:267–310.
- Mehta A, Prabhakar, Prabhakar M, et al. Excitotoxicity: bridge to various triggers in neurodegenerative;disorders. Eur J Pharmacol. 2013;698(1–3):6–18.
- Stone DK, Reynolds AD, Mosley RL, et al. Innate and adaptive immunity for the pathobiology of Parkinson’s disease. Antioxid Redox Signal. 2009 Sep;11(9):2151–2166.
- Ansoleaga B, Jove M, Schluter A, et al. Deregulation of purine metabolism in Alzheimer’s disease. Neurobiol Aging. 2015 Jan;36(1):68–80.
- Blum D, Martire A, Burnouf S, et al. Adenosine Receptors in Huntington’s Disease. New York: Springer New York; 2013. (S. Masino, Boison D, editors. Adenosine: A Key Link between Metabolism and Brain Activity).
- Lopes LV, Sebastiao AM, Ribeiro JA. Adenosine and related drugs in brain diseases: present and future in clinical trials. Curr Top Med Chem. 2011;11(8):1087–1101.
- Chen JF, Huang Z, Ma J, et al. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999 Nov 1;19(21):9192–9200.
- Yu L, Huang Z, Mariani J, et al. Selective inactivation or reconstitution of adenosine A2A receptors in bone marrow cells reveals their significant contribution to the development of ischemic brain injury. Nat Med. 2004 Oct;10(10):1081–1087.
- Srivaratharajah K, Cui A, McAneney J, et al. Chronic hypoxic hypercapnia modifies in vivo and in vitro ventilatory chemoreflexes in the cane toad. Respir Physiol Neurobiol. 2008 Feb 29;160(3):249–258.
- Chalah A, Khosravi-Far R. The mitochondrial death pathway. Adv Exp Med Biol. 2008;615:25–45.
- Calissano P, Matrone C, Amadoro G. Apoptosis and in vitro Alzheimer disease neuronal models. Commun Integr Biol. 2009;2(2):163–169.
- Melah KE, Lu SY-F, Hoscheidt SM, et al. Cerebrospinal fluid markers of alzheimer’s disease pathology and microglial activation are associated with altered white matter microstructure in asymptomatic adults at risk for alzheimer’s disease. J Alzheimers Dis. 2016;50(3):873–886.
- Silva CG, Porciuncula LO, Canas PM, et al. Blockade of adenosine A(2A) receptors prevents staurosporine-induced apoptosis of rat hippocampal neurons. Neurobiol Dis. 2007 Aug;27(2):182–189.
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001 Mar 1;410(6824):37–40.
- Schulte G, Fredholm BB. Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal. 2003 Sep;15(9):813–827.
- Kapfhamer D, Taylor S, Zou ME, et al. Taok2 controls behavioral response to ethanol in mice. Genes Brain Behav. 2013 Feb;12(1):87–97.
- Bachstetter AD, Xing B, Almeida LD, et al. Microglial p38α MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Aβ). J Neuroinflammation. 2011;8(1):79.
- Owens TW, Valentijn AJ, J-P U, et al. Apoptosis commitment and activation of mitochondrial Bax during anoikis is regulated by p38MAPK. Cell Death Differ. 2009;16(11):1551–1562.
- Melani A, Gianfriddo M, Vannucchi MG, et al.The selective A2A receptor antagonist SCH 58261 protects from neurological deficit, brain damage and activation of p38 MAPK in rat focal cerebral ischemia. Brain Res. 2006 Feb 16; 1073–1074:470–480.