
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Maltose phosphorylase (MP), a glycoside hydrolase family 65 enzyme, reversibly phosphorolyzes maltose. In this study, we characterized Bacillus sp. AHU2001 MP (MalE) that was produced in Escherichia coli. The enzyme exhibited phosphorolytic activity to maltose, but not to other α-linked glucobioses and maltotriose. The optimum pH and temperature of MalE for maltose-phosphorolysis were 8.1 and 45°C, respectively. MalE was stable at a pH range of 4.5–10.4 and at ≤40°C. The phosphorolysis of maltose by MalE obeyed the sequential Bi–Bi mechanism. In reverse phosphorolysis, MalE utilized d-glucose, 1,5-anhydro-d-glucitol, methyl α-d-glucoside, 2-deoxy-d-glucose, d-mannose, d-glucosamine, N-acetyl-d-glucosamine, kojibiose, 3-deoxy-d-glucose, d-allose, 6-deoxy-d-glucose, d-xylose, d-lyxose, l-fucose, and l-sorbose as acceptors. The kcat(app)/Km(app) value for d-glucosamine and 6-deoxy-d-glucose was comparable to that for d-glucose, and that for other acceptors was 0.23–12% of that for d-glucose. MalE synthesized α-(1→3)-glucosides through reverse phosphorolysis with 2-deoxy-d-glucose and l-sorbose, and synthesized α-(1→4)-glucosides in the reaction with other tested acceptors.
Graphical Abstract
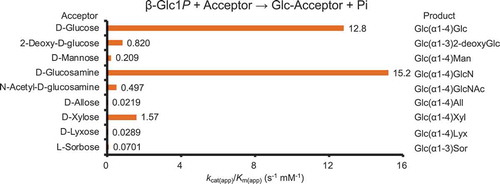
Acceptor specificity of reverse phosphorolysis by MalE and structure of oligosaccharides synthesized.
Maltose phosphorylase (MP, EC 2.4.1.8) catalyzes the reversible phosphorolysis of maltose to d-glucose and β-d-glucose 1-phosphate (β-Glc1P). This enzyme cleaves the glucosidic linkage via the single displacement mechanism, in which the general acid catalyst donates a proton to the scissile glucosidic oxygen, and the inorganic phosphate attacks the anomeric carbon of the glucosyl residue in subsite −1 [Citation1]. Some bacteria such as Bacillus subtilis and Lactobacillus acidophilus utilize MP along with α-glucosidase (EC 3.2.1.20) and neopullulanase (EC 3.2.1.135) for the intracellular metabolism of maltodextrin [Citation2,Citation3]. By utilizing the specific phosphorolytic activity of MP to maltose, quantification methods for maltose and inorganic phosphate were established [Citation4,Citation5].
Based on the amino acid sequence classification of glycoside hydrolases [Citation6], MP is classified into the glycoside hydrolase family 65 (GH65), which is mainly comprised of α-glucoside phosphorylases such as trehalose phosphorylase (EC 2.4.1.64) and kojibiose phosphorylase (EC 2.4.1.230). The GH65 enzymes have an (α/α)6-barrel catalytic domain similar to those of GH15 inverting glycosidases, including glucoamylase (EC 3.2.1.3) and glucodextranase (EC 3.2.1.70) [Citation1,Citation7,Citation8]. GH65 and GH15 enzymes form the clan GH-L. The conserved Glu residue on the loop connecting the fifth and sixth α-helices of the catalytic domain serves as the general acid catalyst of these enzymes [Citation1,Citation7–Citation10]. In GH65 phosphorylases, inorganic phosphate is situated at the position, corresponding to the general base catalyst Glu residue of GH15 glycosidases [Citation1,Citation9,Citation10]. Although the structure of MP in complex with maltose is not available, structural comparison between L. acidophilus MP and Thermoanaerobacterium thermosaccharolyticum glucoamylase suggested that His413 and Glu415 on loop 3, that connects the third and fourth α-helices of the (α/α)6-catalytic domain, are involved in the binding to the reducing end d-glucose residue of maltose in subsite +1 [Citation11].
During reverse phosphorolysis, a sugar phosphate and a sugar serve as the donor and acceptor substrate, respectively. Reverse phosphorolysis is an efficient reaction to synthesize the oligosaccharides [Citation12–Citation18]. Glycoside phosphorylases are generally highly specific to the glycosidic linkage of the substrates and can utilize several sugars as acceptor substrate [Citation3,Citation19–Citation23]. MP can synthesize several non-naturally occurring α-(1→4)-glucosides [Citation16,Citation24]. Vigsnaes et al. reported that these α-(1→4)-glucosides enhanced the growth of Bifidobacterium animalis subsp. lactis and Bifidobacterium longum [Citation25]. Furthermore, MP can also synthesize β-Glc1P from maltose, which can be easily produced from abundant starch. By coupling the reaction of MP and other α-glucoside phosphorylases, several oligosaccharides are produced from maltose. Trehalose and nigerose were synthesized by the coupling reaction of MP and trehalose phosphorylase [Citation13] and MP and nigerose phosphorylase (EC 2.4.1.279) [Citation17], respectively. Although MPs were found in various bacterial origins such as Bacillus selenitireducens [Citation26], Bacillus sp. RK-1 [Citation27], Enterococcus faecalis [Citation28], Lactobacillus acidophilus [Citation3], Lactobacillus brevis [Citation29], Lactobacillus sanfranciscensis [Citation30], Lactobacillus sp [Citation31]., Paenibacillus sp. SH-35 [Citation32], and Propionibacterium freudenreichii [Citation33], acceptor specificity of MP in the reverse phosphorolysis was analyzed using only a few MPs [Citation3,Citation26,Citation33]. The MPs show relatively high activity of reverse phosphorolysis to acceptor of d-glucose derivatives at the 2-C and 6-C positions, including d-glucosamine, N-acetyl-d-glucosamine, and d-xylose, and form α-(1→4)-glucosides.
The gene malE that encodes GH family 65 enzyme is downstream of the GH31 α-glucosidase gene (BspAG31A) of Bacillus sp. AHU2001 (formerly known as Bacillus sp. SW20) [Citation34]. The deduced amino acid sequence of MalE share 52–60% identity with that of characterized MPs: B. selenitireducens MP (GenBank number, ADH99560.1); Bacillus sp. RK-1 MP (GenBank number, BAC54904.1); E. faecalis MP (GenBank number, AAO80764.1); L. acidophilus MP (GenBank number, AAV43670.1); L. brevis MP (Uniprot number, Q7SIE1); L. sanfranciscensis MP (GenBank number, ADH99560.1); and Paenibacillus sp. SH-35 MP (GenBank number, BAD97810.1). In this study, we characterized a recombinant MalE that was produced in Escherichia coli, and described acceptor specificity of this enzyme in detail to advance understanding of acceptor specificity of MP. Kinetic analysis of the reverse phosphorolysis was performed with not only good acceptor substrates but also poor acceptors, and the general kinetic model for the reaction with sugar phosphate and an acceptor substrate, in which hydrolysis of β-Glc1P also occurred along with the reverse phosphorolysis, was proposed. Furthermore, the chemoenzymatic synthesis of oligosaccharides using MalE was established.
Materials and methods
Chemicals
We used the following sugars as substrate: d-glucose, 1,5-anhydro-d-glucitol, methyl α-d-glucoside, methyl β-d-glucoside, d-mannose, kojibiose, d-allose, nigerose, d-xylose, d-glucuronic acid, d-tagatose (Wako Pure Chemical Industries; Osaka, Japan); 2-deoxy-d-glucose, d-glucosamine, isomaltose (Tokyo Chemical Industry; Tokyo, Japan); 3-deoxy-d-glucose, 4-deoxy-d-glucose (Carbosynth, Berkshire, UK); 6-deoxy-d-glucose, l-fucose, d-talose, l-rhamnose, d-altrose, d-xylulose, (Sigma, St. Louis, MO, USA); maltotriose, N-acetyl-d-glucosamine, d-galactose, d-fructose, l-sorbose (Nacalai Tesque; Kyoto, Japan); d-lyxose (Alfa Aesar, Heysham, Lancashire, UK); l-arabinose (Kanto Chemical, Tokyo, Japan); trehalose (Hayashibara, Okayama, Japan); and maltose (Nihon Shokuhin Kako, Tokyo, Japan).
Preparation of recombinant MalE
The expression plasmid derived from pET-23a (Novagene, Darmstadt, Germany) was prepared to obtain a recombinant MalE with eight extra amino acid residues (Leu-Glu-His-His-His-His-His-His), attached to the C-terminal of MalE (GenBank accession number: BAQ19546.1). The NdeI and XhoI restriction sites were introduced to the 5ʹ- and 3ʹ-terminals of malE, respectively. An internal NdeI site was mutated by the overlap extension polymerase chain reaction (PCR) method [Citation35]. In the first PCR, the 5ʹ- and 3ʹ-regions of malE were amplified from the genomic DNA of Bacillus sp. AHU2001 using the following two sets of primers: for the 5ʹ-region, 5ʹ-AAGGTGATAACATATGAAAAGATTAT-3ʹ (primer A, sense orientation, NdeI site underlined) and 5ʹ-CGCATCCCCATGTGACCGTTTCC-3ʹ (antisense orientation, substituted nucleotide in boldface); for the 3ʹ-region, 5ʹ-GGAAACGGTCACATGGGGATGCG-3ʹ (sense orientation) and 5ʹ-AGCTTCATATCTCGAGGCCGGAATTGCTTG-3ʹ (primer B, antisense, XhoI site underlined). The second PCR was performed using the amplified DNA fragments as template and primers A and B. The amplified DNA fragment and pET-23a were subjected to restriction digestion using NdeI and XhoI (Takara Bio, Kusatsu, Japan). The resulting DNA fragments were ligated using DNA Ligation Kit Mighty Mix (Takara Bio). The DNA sequence of the insert and the flanking regions was analyzed using an Applied Biosystems 3130 Genetic Analyzer (Life Technologies, Carlsbad, CA, USA).
Recombinant MalE was produced in the transformant of E. coli BL21 (DE3), harboring the expression plasmid. The transformant was incubated in 2 L Luria-Bertani (LB) medium, containing 100 μg/mL ampicillin, at 37°C with vigorous shaking until the culture absorbance at 600 nm (A600) reached 0.5. The recombinant protein production was induced by adding 2 mL of 100 mM isopropyl β-d-thiogalactoside to the culture medium (final concentration, 0.1 mM), and the cells were further incubated at 18°C for 22 h. The bacterial cells were collected by centrifugation (8000 × g, 4°C, 10 min), and were disrupted in 100 mL of 30 mM imidazole-HCl buffer (pH 6.5) containing 500 mM NaCl by sonication. The cell debris was removed by centrifugation (12,000 × g, 4°C, 10 min) and the cell-free extract was obtained as the starting material for MalE purification. The proteins were applied to a Ni2+-immobilized Chelating Sepharose column (2.8 cm i.d. × 5 cm; GE Healthcare, Uppsala, Sweden). The column was washed with the same buffer and the adsorbed protein was eluted with a linear gradient of imidazole from 30 to 500 mM (total elution volume, 400 mL). The collected fractions, containing highly purified MalE, were pooled and dialyzed against 10 mM 2-morpholinoethanesulfonic acid (MES)-NaOH buffer (pH 6.5).
Protein quantification
During the purification process, the protein concentration was determined by the UV method [Citation36]. The absorbance was measured at 280 nm (A280) to determine the protein concentration. The protein concentration (mg/mL) was assumed to be equal to A280. The concentration of purified protein was determined by the amino acid analysis using JLC-500/V (JEOL, Tokyo, Japan) after hydrolysis of the purified protein in 6 M HCl at 110°C for 24 h.
Measurement of molecular mass by gel filtration column chromatography
The molecular mass of MalE in the native state was measured by gel filtration column chromatography under the following conditions: column, Superdex 200 10/300 GL (GE Healthcare); elution, 10 mM 2- [4-(2-hydroxyethyl)-1-piperazinyl]ethanesulfonic acid (HEPES)-NaOH buffer (pH 6.5) containing 0.2 M NaCl; flow rate, 0.5 mL/min; detection, A280. A set of gel filtration standards (Bio-Rad, Hercules, CA, USA) was used to calibrate the molecular mass.
Preparation of β-Glc 1P
β-Glc 1P was prepared by the phosphorolysis of maltose using MalE. A reaction mixture (2 L), containing 0.3 M maltose (600 mmol), 0.3 M potassium phosphate buffer (pH 8.0; 600 mmol), and 16.5 mg/L MalE, was incubated at 37°C for 96 h. From the reaction, 178 mmol β-Glc 1P was obtained. The pH of the reaction mixture was adjusted to pH 4.0 with 6 M HCl. Further, 4 mL glucoamylase (AMG300L; Novozymes, Bagsværd, Denmark) was added, and the mixture was incubated at 60°C for 3 h to digest the remaining maltose. The pH of the mixture was adjusted to pH 6.5 with 5 M KOH. Next, 41 mL of 28% (w/v) ammonium water and 128 g magnesium acetate were added and the mixture was incubated at 4°C for 1 h. The insoluble NH4MgPO4 · 6H2O was removed by filtration using Celite No. 545 (Wako Pure Chemical Industries). The resulting solution was concentrated to 500 mL under reduced pressure. The pH of the solution was adjusted to pH 8.5 with 5 M KOH, and the insoluble NH4MgPO4 · 6H2O was removed as described above. Methanol (2.4 L) was added to the mixture and incubated at 25°C for 2 h. The precipitate was collected by filtration using a filter paper, and dissolved in 700 mL water. The yield of β-Glc1P obtained was 164 mmol. Five hundred milliliter of this mixture containing 117 mmol β-Glc1P was further subjected to anion exchange column chromatography with Amberjet 4400 (3.6 cm i.d. × 90 cm, acetate form; Organo, Tokyo, Japan) according to a method previously described [Citation37]. The fractions containing β-Glc1P were concentrated under reduced pressure, and β-Glc1P was precipitated in 80% ethanol. β-Glc1P, collected by filtration, was dried in vacuo, and stored at −20°C. The yield of β-Glc1P dipotassium salt obtained was 72.8 mmol.
Standard enzyme assay
A reaction mixture (50 μL), containing appropriate concentration of enzyme (2.94–14.7 μg/mL), 4 mM maltose, 10 mM sodium phosphate buffer (pH 8.0), 100 mM HEPES-NaOH buffer (pH 8.0), and 0.2 mg/mL bovine serum albumin (BSA), was incubated at 37°C for 10 min. The reaction was stopped by heating at 90°C for 5 min. One hundred microliter of 2 M Tris-HCl buffer (pH 7.0) was added to the sample, and the released d-glucose was measured by the d-glucose oxidase-peroxidase method [Citation38] using glucose CII test (Wako Pure Chemical Industries). One unit (U) of MP activity was defined as the amount of enzyme required to phosphorolyze 1 μmol of maltose in 1 min under these conditions.
Evaluation of the effect of pH and temperature on enzyme activity and stability
The optimal pH for enzyme activity was measured under the standard assay conditions but 100 mM HEPES-NaOH buffer (pH 8.0) was replaced with 100 mM of various buffers: sodium citrate buffer (pH 4.1–6.1), MES-NaOH buffer (pH 6.1–7.1), HEPES-NaOH buffer (pH 7.1–8.5), and glycine-NaOH buffer (pH 8.6–9.6). The optimal temperature for the enzyme activity was determined at various temperatures. The pH stability was evaluated by determining the residual enzyme activity after incubating 0.29 mg/mL MalE with 100 mM Britton Robinson buffer (pH 3.5–11.3) at 4°C for 24 h. The treated enzyme solution was diluted by 20 times with 10 mM HEPES-NaOH buffer (pH 8.0) containing 1 mg/mL BSA for the activity assay. The stable range for temperature was evaluated by measuring the residual enzyme activity after incubating 15 μg/mL MalE with 100 mM sodium phosphate buffer (pH 8.0) at 30–60°C for 15 min.
Kinetic analysis of the phosphorolysis of maltose
The reaction rate for phosphorolysis of maltose at various concentrations of maltose (0.5–4 mM) and sodium phosphate buffer (1–8 mM) was measured as per the standard enzyme assay. The kinetic parameters were calculated by fitting the reaction equation Equation (1) for a sequential Bi–Bi mechanism [Citation39] to the reaction rates with Grafit version 7.0.2 (Erithacus Software, East Grinstead, UK).
where A = inorganic phosphate, B = maltose
Substrate specificity
The phosphorolytic activity of the recombinant protein was measured using 4 mM maltotriose, kojibiose, nigerose, isomaltose, and trehalose under the standard assay conditions.
To investigate the acceptor specificity in the reverse phosphorolysis, inorganic phosphate releasing velocity to β-Glc1P and acceptors (monosaccharides and disaccharides mentioned above) was measured. A reaction mixture (20 μL), containing 1.47–588 μg/mL MalE, 10 mM β-Glc1P, 10 mM acceptor, and 100 mM HEPES-NaOHbuffer (pH 8.0), was incubated at 37°C for 10 min. The sample was heated at 90°C for 5 min to stop the reaction, and the released inorganic phosphate was measured following the methods of Lowry and Lopez [Citation40].
To determine the kinetic parameters for the reaction to β-Glc1P and acceptor, we used a reaction scheme that involves both the hydrolysis of β-Glc1P and the reverse phosphorolysis (). The rate equation Equation (2), obtained from the scheme was as follows:
Figure 1. Kinetic scheme of the reverse phosphorolysis.
where [P] and [Q] are molar concentration of β-Glc1P and acceptor, respectively.
Kinetic parameters, KmP, KmQ, and Ks, were defined as follows by the rate constants shown in :
At a constant [P], the rate equation is shown as Equation (3).
v0 is the hydrolytic velocity of β-Glc1P without the acceptor:
The apparent kinetic parameters, kcat(app) and Km(app), are given by the following equations:
The apparent kinetic parameters for d-glucose, 1,5-anhydro-d-glucitol, methyl α-d-glucoside, 2-deoxy-d-glucose, d-mannose, d-glucosamine, N-acetyl-d-glucosamine, kojibiose, 3-deoxy-d-glucose, d-allose, 6-deoxy-d-glucose, d-xylose, d-lyxose, l-fucose, and l-sorbose, were determined by fitting the reaction equation Equation (3) to reaction rates at 0.75‒25 mM of the acceptor and 10 mM β-Glc1P.
kcat1 and Ks, which are the kinetic parameters for the hydrolysis of β-Glc1P, were determined by fitting Equation (4) to the hydrolytic velocity to various concentrations of β-Glc1P in the absence of acceptor.
Preparation and structural analysis of oligosaccharides produced by the reverse phosphorolysis of MalE
A reaction mixture (1 or 50 mL), containing 8.81–145 μg/mL MalE, 0.1 M β-Glc1P, 0.1 M acceptor (d-glucosamine, 2-deoxy-d-glucose, d-mannose, N-acetyl-d-glucosamine, d-allose, d-xylose, d-lyxose, or l-sorbose), and 0.1 M HEPES-NaOH buffer (pH 8.0), was incubated at 37°C for 5–72 h. For the reaction with d-allose, 2-deoxy-d-glucose, d-lyxose, and l-sorbose, 0.1 M magnesium acetate and 0.1 M ammonium acetate were added, and the insoluble NH4MgPO4 · 6H2O was removed by centrifugation after the reaction. The synthesized oligosaccharides were purified by gel filtration column chromatography using Bio-Gel P2 [Bio-Rad; 1.6 cm i.d. × (60 cm + 60 cm + 100 cm)], and lyophilized. The molecular mass of the products was measured by electrospray ionization mass spectrometry (ESI-MS) using an Exactive mass spectrometer (Thermo Scientific, San Jose, CA, USA). The sample was applied to the mass spectrometer by flow injection. Methanol was used as the mobile phase solvent. The positive ion was detected under the following conditions: spray voltage, 3.00 kV; capillary temperature, 300°C. NMR spectra were recorded in D2O (Sigma) at 27°C using a Bruker AMX500 (500 MHz; Bruker Corporation, Billerica, MA, USA). A series of two-dimensional homo- and heteronuclear correlated spectra [correlated spectroscopy, heteronuclear single quantum correlation (HSQC) spectroscopy, HSQC total correlation spectroscopy, heteronuclear 2 bond correlation spectroscopy, and heteronuclear multiple bond correlation spectroscopy] were acquired to determine the chemical structure of the reaction products.
Digestion of the synthesized oligosaccharides using rat intestinal α-glucosidase
The digestion rate of the oligosaccharides obtained from 2-deoxy-d-glucose, d-mannose, d-glucosamine, N-acetyl-d-glucosamine, d-allose, d-xylose, d-lyxose, and l-sorbose through the reverse phosphorolysis was compared with that of maltose and isomaltose. To prepare the α-glucosidase, 13 mg of rat intestine acetone powder (Sigma) was suspended in 0.26 mL of 10 mM sodium phosphate buffer (pH 6.5), and the supernatant was collected by centrifugation (13,000 × g, 4°C, 5 min). A reaction mixture (50 μL), containing α-glucosidase, 10 mM glucoside, and 40 mM sodium phosphate buffer (pH 6.5), was incubated at 37°C for 10 min. The reaction was terminated by adding 100 μL of 2 M Tris-HCl buffer (pH 7.0), and the released d-glucose was measured as described above.
Results
Preparation and basic properties of MalE
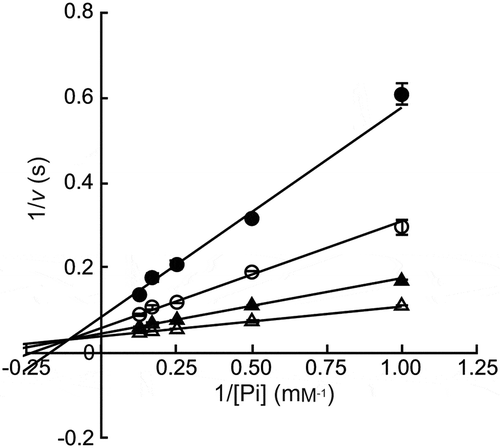
The recombinant protein, MalE was successfully produced in E. coli BL21 (DE3) transformant, and purified to homogeneity by nickel affinity column chromatography. The yield of the purified enzyme from the cells obtained from 2 L culture broth was 13.6 mg and a specific activity was 15.9 U/mg. The molecular mass of MalE was 90 kDa as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; theoretical molecular mass is 89.6 kDa; Figure. S1) and 193 kDa as determined by gel filtration column chromatography. This indicated that MalE exists as homodimer under nondenaturing conditions similar to other MPs [Citation3,Citation26]. MalE exhibited the highest activity at pH 8.1 and at 45°C (). The residual activity was higher than 80% of the original activity at a pH range of 4.5–10.4 after the pH treatment at 4°C for 24 h and at temperatures ≤40°C after the heat treatment at pH 8.0 for 15 min. During the phosphorolysis of maltose, the reaction equation for a sequential Bi–Bi mechanism fitted well to the reaction rate at various maltose and phosphate concentrations (): kcat, 30.9 ± 0.6 s−1; KmA, 0.295 ± 0.059 mM; KmB, 0.835 ± 0.123 mM; and KiA, 9.07 ± 1.74 mM (A, phosphate; B, maltose). We did not detect any phosphorolytic activity of MalE to maltotriose, kojibiose, nigerose, isomaltose, and trehalose (less than 0.6% of that to maltose).
Figure 2. Effects of pH and temperature.
(a) pH activity curve. Reaction buffers are sodium citrate buffer (open circles), MES-NaOH buffer (close circles), HEPES-NaOH buffer (open squares), and glycine-NaOH buffer (close squares). (b) Temperature activity curve. (c) pH stability. Residual activity after the pH treatment at 4°C for 24 h is shown. (d) Temperature stability. Residual activity after the heat treatment for 15 min is shown. Values and error bars are average and standard deviation of three independent experiments.
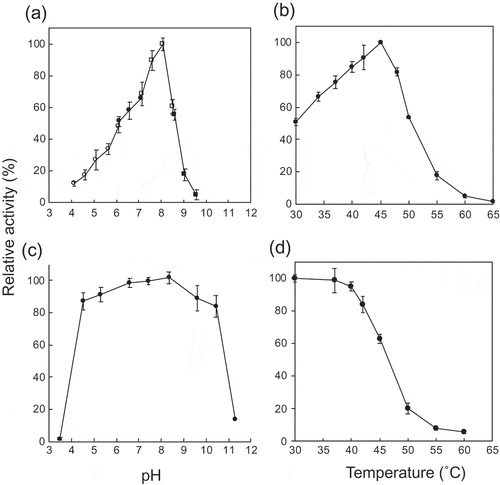
Figure 3. Double reciprocal plots for phosphorolysis of maltose by MalE.
Maltose concentrations were 0.5 mM (closed circles), 1.0 mM (open circles), 2.0 mM (closed triangles), and 4.0 mM (open triangles). Values and error bars are average and standard deviation of three independent experiments.
Acceptor specificity of MalE in reverse phosphorolysis
The acceptor specificity of MalE was investigated based on the inorganic phosphate releasing velocity to 30 types of sugar and β-Glc1P. Compared to the reaction without acceptor, the production of inorganic phosphate was enhanced by the addition of d-glucose, 1,5-anhydro-d-glucitol, methyl α-d-glucoside, 2-deoxy-d-glucose, d-mannose, d-glucosamine, N-acetyl-d-glucosamine, kojibiose, 3-deoxy-d-glucose, d-allose, 6-deoxy-d-glucose, d-xylose, d-lyxose, l-fucose, and l-sorbose as acceptors. Such enhancement was not observed in the reactions with methyl β-d-glucoside, trehalose, nigerose, 4-deoxy-d-glucose, d-galactose, maltose, d-glucuronic acid, isomaltose, d-talose, l-rhamnose, d-altrose, l-arabinose, d-fructose, d-tagatose, and d-xylulose, indicating that these sugars did not serve as acceptors. The apparent kinetic parameters for the reverse phosphorolysis with the acceptors were determined based on the inorganic phosphate releasing velocity in the presence of 10 mM β-Glc1P (). During the reverse phosphorolysis with good acceptors, the hydrolysis of β-Glc1P can be neglected, and the apparent kinetic parameters for acceptor can be calculated by fitting the Michaelis-Menten equation to the reaction velocity at various concentrations of acceptor and a constant concentration of sugar phosphate. However, the hydrolytic activity of β-Glc1P was not negligible in the reactions to 1,5-anhydro-d-glucitol, methyl α-d-glucoside, d-allose, d-lyxose, l-fucose, and l-sorbose due to low activity of reverse phosphorolysis (Figure. S2). Thus, the reaction equation Equation (3) obtained from the reaction scheme, in which both hydrolysis of β-Glc1P and the reverse phosphorolysis occur, was employed to determine the apparent kinetic parameters. The reaction velocity for 10 mM β-Glc1P (v0) was calculated to be 0.253 s−1 from the kinetic parameters of β-Glc1P hydrolysis in the absence of acceptors: kcat1, 0.983 ± 0.029 s−1; and Ks, 28.9 ± 1.1 mM. The inorganic phosphate releasing velocity for all the tested acceptors fitted well to Equation (3) (Figure. S2). The apparent kinetic parameters were determined as shown in . The apparent kinetic parameters for d-glucose, 2-deoxy-d-glucose, d-mannose, d-glucosamine, N-acetyl-d-glucosamine, kojibiose, 3-deoxy-d-glucose, 6-deoxy-d-glucose, and d-xylose, determined using Equation (3), were almost consistent with the values obtained by the Michaelis-Menten equation (data not shown), because the velocity for β-Glc1P hydrolysis was much lower than the velocity for the reverse phosphorolysis. MalE exhibited high kcat(app)/Km(app) for d-glucose, d-glucosamine, and 6-deoxy-d-glucose (12.8, 15.2, and 12.2 s−1 mM−1, respectively). The kcat(app)/Km(app) value for the other acceptors was 0.23–12% of that to d-glucose, because of lower kcat(app) and higher Km(app) values.
Table 1. Apparent kinetic parameters for reverse phosphorolysis with various acceptor substrates.
Synthesis of oligosaccharides through reverse phosphorolysis
The chemical structure of oligosaccharides produced by the reverse phosphorolysis with 2-deoxy-d-glucose, d-mannose, d-glucosamine, N-acetyl-d-glucosamine, d-allose, d-xylose, d-lyxose, and l-sorbose was examined. In the reaction with d-mannose, d-glucosamine, N-acetyl-d-glucosamine, and d-xylose (starting substrates: 5 mmol of β-Glc1P and acceptor), the obtained amount of the oligosaccharides after the purification was 1.3 mmol (26% yield), 0.66 mmol (13% yield), 2.0 mmol (39% yield), and 2.4 mmol (49% yield), respectively. The yield of oligosaccharides from 100 μmol of β-Glc1P and acceptors (2-deoxy-d-glucose, d-allose, d-lyxose, and l-sorbose) was 38 μmol (38% yield), 33 μmol (33% yield), 61 μmol (61% yield), and 76 μmol (76% yield), respectively. The chemical structure of the oligosaccharides prepared was analyzed by ESI-MS and NMR. The molecular mass of the reaction products from 2-deoxy-d-glucose, d-mannose, d-glucosamine, N-acetyl-d-glucosamine, d-allose, d-xylose, d-lyxose, and l-sorbose was 349 m/z [M + Na]+, 365 m/z [M + Na]+, 342 m/z [M + H]+, 406 m/z [M + Na]+, 365 m/z [M + Na]+, 335 m/z [M + Na]+, 335 m/z [M + Na]+, and 381 m/z [M + K]+, respectively, indicating that all the oligosaccharides were disaccharides. The NMR analysis revealed that the reaction products from d-mannose, d-glucosamine, N-acetyl-d-glucosamine, d-allose, d-xylose, and d-lyxose were α-(1→4)-glucosides, and the products from 2-deoxy-d-glucose and l-sorbose were α-(1→3)-glucosides ().
Table 2. Chemical shifts of oligosaccharides produced through the reverse phosphorolysis in the 1H- and 13C- NMR spectra.
Digestion of the oligosaccharides by rat intestinal α-glucosidase
The digestion rate of the oligosaccharides from 2-deoxy-d-glucose, d-mannose, d-glucosamine, N-acetyl-d-glucosamine, d-allose, d-xylose, d-lyxose and l-sorbose using rat intestinal α-glucosidase was compared with that of maltose and isomaltose. The digestion rate of Glc(α1-3)2-deoxyGlc, Glc(α1-4)Man, Glc(α1-4)GlcN, Glc(α1-4)GlcNAc, Glc(α1-4)All, Glc(α1-4)Xyl, Glc(α1-4)Lys, and Glc(α1-3)Sor was 28.1 ± 0.9%, 43.8 ± 2.5%, 39.8 ± 2.0%, 40.4 ± 0.3%, 37.9 ± 0.8%, 45.0 ± 0.3%, 39.3 ± 1.2%, and 28.0 ± 0.7% of that of maltose, respectively. These digestion rates were higher than that of isomaltose (15.2 ± 0.8%). The tested α-(1→3)-glucosides exhibited higher tolerance against the α-glucosidase than the α-(1→4)-glucosides.
Discussion
MP is involved in the intracellular metabolism of maltodextrin and is useful for the synthesis of oligosaccharides through reverse phosphorolysis [Citation16,Citation24,Citation26]. Previously, we had reported that the MP gene (malE) of Bacillus sp. AHU 2001 was downstream of the gene encoding GH31 α-glucosidase BspAG31A [Citation34]. In this study, we characterized the recombinant MalE, and the enzyme was used to synthesize several oligosaccharides. Maltose was the best substrate for MalE phosphorolysis. Therefore, MalE is probably involved in the intracellular metabolism of maltodextrin along with BspAG31A. MalE did not exhibit any detectable phosphorolytic activity to trehalose, kojibiose, nigerose, and isomaltose, and was specific to α-(1→4)-glucosidic linkage, similar to other MPs. To understand the substrate binding mechanism, we tried to predict the binding mode of d-glucose in subsite +1 of MalE through structural comparison between a model structure of MalE and the crystal structure of Caldicellulosiruptor saccharolyticus kojibiose phosphorylase [Citation9]. Model structure of MalE was constructed through a homology modeling with Phyre2 program [Citation41]. In the model structure of MalE, the orientation of the amino acid residues in the substrate binding site were consistent well with that of the template protein (L. brevis MP [Citation1]; amino acid sequence of MalE is 52% identical with that of L. brevis MP). The superimposition showed that the amino acid residues in subsite −1 of MalE, including the general acid catalyst Glu486, were predicted to be located at similar position to the corresponding residues of C. saccharolyticus kojibiose phosphorylase ()). This suggested that MalE and the kojibiose phosphorylase share the binding mode of non-reducing end d-glucosyl residue, meaning that the glucosidic oxygen (4-O and 2-O of the reducing end d-glucose residue of maltose and kojibiose, respectively) is situated at the same position in these enzymes. To predict the binding mode of maltose, α-d-glucose, which is utilized as acceptor of MP in the reverse phosphorolysis [Citation42], was superimposed onto the reducing end d-glucose residue of kojibiose ()). His415, Glu417, and Lys595 of MalE were predicted to form hydrogen bonds with 5-O, 1-O and 3-O of the superimposed d-glucose in subsite +1, respectively ()). The hydrogen bonds formed by Glu417 and Lys595 are consistent with the prediction using L. acidophilus MP [Citation11]. In addition to these residues, Tyr351 is predicted to form a hydrogen bond with 3-O of d-glucose in subsite +1. As the residues of MalE, predicted to be involved in the formation of subsite +1, are conserved in MPs (Figure. S3), the substrate binding mode is presumably common to MPs.
Figure 4. Predicted substrate binding structure of MalE.
(a) Superimposition of model structure of MalE (magenta) and C. saccharolyticus kojibiose phosphorylase (PDB entry, 3WIQ; green). Glucosyl residue bound to subsite −1 of the kojibiose phosphorylase is shown in cyan. α-d-Glucose (PDB entry, AGC), shown in yellow, is superimposed onto d-glucose residue of kojibiose in subsite +1 (this d-glucose residue is not shown in this panel). Dotted line indicates predicted hydrogen bonds. (b) Superimposition of α-d-glucose (yellow) and the reducing end d-glucose residue of kojibiose. (c) Surface model of MalE. α-d-Glucose bound to subsite +1 is shown in the yellow stick representation. The amino acid residues forming subsite +1 are shown in magenta.
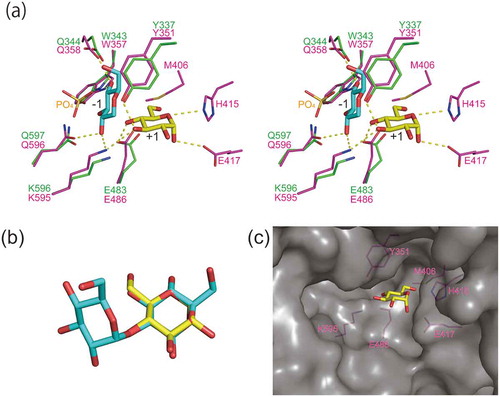
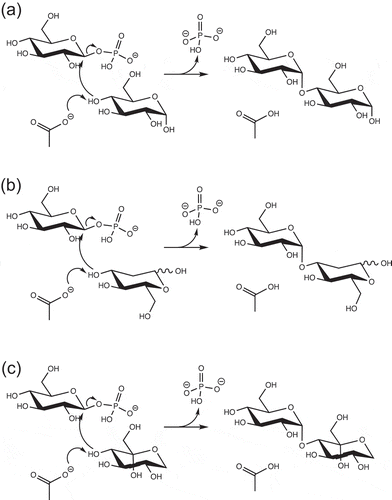
The kcat(app)/Km(app) for reverse phosphorolysis of d-glucosamine by MalE was as high as that of d-glucose, although the kcat(app)/Km(app) values for 2-deoxy-d-glucose and d-mannose were 16- and 61-fold lower than that for d-glucose. This indicated that the equatorial 2-hydroxy group is important for the stabilization of transition state during the reverse phosphorolysis, and that the amino group can replace 2-OH group of d-glucose. MalE also utilized acceptors harboring equatorial bulky chemical group at the 2-C position, N-acetyl-d-glucosamine, as observed in MPs of Bacillus selenitireducens [Citation26], L. acidophilus [Citation16], and Neisseria perflava [Citation24]. Furthermore, MalE utilized kojibiose as an acceptor like B. selenitireducens MP, which glucosylates the reducing end d-glucose residue of kojibiose through the reverse phosphorolysis [Citation26]. MalE could also transfer d-glucosyl residue to the reducing end of kojibiose. The model structure of MalE suggested that MalE does not interact with 2-O of d-glucose in subsite +1, and has open space near the 2-O of this d-glucose ()). This space could account for the activity of reverse phosphorolysis to the derivatives of d-glucose with bulky chemical group at the 2-C position. Although some MPs are reported to glucosylate 2-deoxy-d-glucose [Citation24,Citation26], the structure of the reaction product has not yet been determined. Structural analysis of the product from 2-deoxy-d-glucose revealed that α-(1→3)-glucoside was synthesized, which suggested that 2-deoxy-d-glucose in 3CO may bind to subsite +1 upside down, and that 3-OH group serves as the nucleophile during the reverse phosphorolysis (). The hydroxymethylene group of 2-deoxy-d-glucose could be accommodated in the space described above.
Figure 5. Possible reaction mechanisms of reverse phosphorolysis.
Possible reaction mechanisms of reverse phosphorolysis with α-d-glucose (a), 2-deoxy-d-glucose (b), and α-l-sorbopyranose (c) are shown.
The kcat(app)/Km(app) of MalE for 6-deoxy-d-glucose was comparable with that of d-glucose. However, the kcat(app)/Km(app) of MalE for d-xylose was considerably lower than that for d-glucose. This suggests that 6-C, and not 6-OH, of d-glucose is involved in binding at subsite +1 of MalE. Met406 of MalE, predicted to be situated near the hydroxymethylene group of d-glucose in subsite +1 ()), might interact with the 6-C of d-glucose in subsite +1 through a hydrophobic interaction. In contrast to the reaction with the d-glucose derivatives of 2-C and 6-C positions, MalE exhibited very low synthetic activity to all the 1- and 3-OH d-glucose derivatives tested: 1,5-anhydro-d-glucitol, methyl α-d-glucoside, 3-deoxy-d-glucose, and d-allose, indicating that 1- and 3-hydroxy groups of d-glucose are important for the substrate binding. Tyr351, Glu417, and Lys595, predicted to interact with these hydroxy groups, are thought to be essential for the substrate binding. The importance of these hydroxy groups of the acceptor substrate for the reverse phosphorolysis is also reported in MPs from other origins [Citation3,Citation26,Citation33].
MalE exhibited weak but apparent reverse phosphorolysis activity to a ketose, l-sorbose, and catalyzed the formation of α-(1→3)-glucoside, indicating that 3-O of l-sorbose acted as the nucleophile in the reverse phosphorolysis. As 1-C, 1-O, 2-C, 3-C, 3-O, 4-C, 4-O, 5-C, 5-O, 6-C and 6-O of α-l-sorbopyranose could be located at positions similar to 6-C, 6-O, 5-C, 4-C, 4-O, 3-C, 3-O, 2-C, 2-O, 1-C, and 5-O of d-glucose, respectively, α-l-sorbopyranose presumably served as the acceptor in this reaction ().
Among the oligosaccharides produced by MalE, digestibility of Glc(α1-3)2-deoxyGlc, Glc(α1-4)Man, Glc(α1-4)GlcN, Glc(α1-4)GlcNAc, Glc(α1-4)All, Glc(α1-4)Xyl, and Glc(α1-3)Sor by rat intestinal α-glucosidase was investigated. These oligosaccharides are less digestible compared to maltose. This indicated that these oligosaccharides could reach large intestine. Glc(α1-4)GlcNAc promotes the growth of B. animalis and B. longum [Citation25]. As Glc(α1-3)2-deoxyGlc and Glc(α1-3)Sor exhibited higher tolerance against the α-glucosidase than the α-(1→4)-glucosides, they might also act as prebiotic oligosaccharides.
In this study, we demonstrated the enzymatic functions of MalE and evaluated the oligosaccharides synthesized by this enzyme. Although the substrate binding mechanism was discussed based on the predicted MalE structure, the structure of complex of MP and maltose, which is experimentally determined through X-ray crystallography, is required to clarify the structural-function relationship of this typical GH65 enzyme.
Author contribution
Wataru Saburi conceived and designed the experiments. Yu Gao, Wataru Saburi, and Yodai Taguchi carried out the experiments. Yu Gao, Wataru Saburi and Haruhide Mori described the manuscript. Haruhide Mori provided overall supervision of the study. All authors have read and approved the final manuscript of this study.
Supplemental_material.pptx
Download MS Power Point (6.4 MB)Acknowledgments
We thank Dr. Eri Fukushi from the GC-MS & NMR Laboratory, Research Faculty of Agriculture, Hokkaido University for NMR data analysis, Ms. Nozomi Takeda and Mr. Tomohiro Hirose of the Global Facility Center, Hokkaido University for the amino acid and MS analyses, and Mr. Yusuke Takada from the DNA sequencing facility of the Research Faculty of Agriculture, Hokkaido University for assistance with DNA sequence analysis.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Egloff MP, Uppenberg J, Haalck L, et al. Crystal structure of maltose phosphorylase from Lactobacillus brevis: unexpected evolutionary relationship with glucoamylases. Structure. 2001;9:689–697.
- Schönert S, Seitz S, Krafft H, et al. Maltose and maltodextrin utilization by Bacillus subtilis. J Bacteriol. 2006;188:3911–3922.
- Nakai H, Baumann MJ, Petersen BO, et al. The maltodextrin transport system and metabolism in Lactobacillus acidophilus NCFM and production of novel α-glucosides through reverse phosphorolysis by maltose phosphorylase. FEBS J. 2009;276:7353–7365.
- Kamogawa A, Yokobayashi K, Fukui T. An enzymatic method for the determination of maltose in the presence of other oligosaccharides. Anal Biochem. 1974;57:303–305.
- Hüwel S, Haalck L, Conrath N, et al. Maltose phosphorylase from Lactobacillus brevis: purification, characterization, and application in a biosensor for ortho-phosphate. Enzyme Microb Technol. 1997;21:413–420.
- Lombard V, Golaconda Ramulu H, Drula E, et al. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495.
- Aleshin A, Golubev A, Firsov LM, et al. Crystal structure of glucoamylase from Aspergillus awamori var. X100 to 2.2-Å resolution. J Biol Chem. 1992;267:19291–19298.
- Mizuno M, Tonozuka T, Suzuki S, et al. Structural insights into substrate specificity and function of glucodextranase. J Biol Chem. 2004;279:10575–10583.
- Okada S, Yamamoto T, Watanabe H, et al. Structural and mutational analysis of substrate recognition in kojibiose phosphorylase. FEBS J. 2014;281:778–786.
- Touhara KK, Nihira T, Kitaoka M, et al. Structural basis for reversible phosphorolysis and hydrolysis reactions of 2-O-α-glucosylglycerol phosphorylase. J Biol Chem. 2014;289:18067–18075.
- Nakai H, Petersen BO, Westphal Y, et al. Rational engineering of Lactobacillus acidophilus NCFM maltose phosphorylase into either trehalose or kojibiose dual specificity phosphorylase. Protein Eng Des Sel. 2010;23:781–787.
- Kitaoka M, Sasaki T, Taniguchi H. Conversion of sucrose into cellobiose using sucrose phosphorylase, xylose isomerase and cellobiose phosphorylase. Denpun Kagaku. 1992;39:281–283.
- Yoshida M, Nakamura N, Horikoshi K. Production of trehalose by a dual enzyme system of immobilized maltose phosphorylase and trehalose phosphorylase. Enzyme Microbial Technol. 1998;22:71–75.
- Chaen H, Nishimoto T, Nakada T, et al. Enzymatic synthesis of kojioligosaccharides using kojibiose phosphorylase. J Biosci Bioeng. 2001;92:177–182.
- Nishimoto M, Kitaoka M. Practical preparation of lacto-N-biose I, a candidate for the bifidus factor in human milk. Biosci Biotechnol Biochem. 2007;71:2101–2104.
- Nakai H, Dilokpimol A, Abou Hachem M, et al. Efficient one-pot enzymatic synthesis of α-(1→4)-glucosidic disaccharides through a coupled reaction catalysed by Lactobacillus acidophilus NCFM maltose phosphorylase. Carbohydr Res. 2010;345:1061–1064.
- Nihira T, Miyajima F, Chiku K, et al. One pot enzymatic production of nigerose from common sugar resources employing nigerose phosphorylase. J Appl Glycosci. 2014;61:75–80.
- Abe K, Nakajima M, Kitaoka M, et al. Large-scale preparation of 1,2-β-glucan using 1,2-β-oligoglucan phosphorylase. J Appl Glycosci. 2015;62:47–52.
- Taguchi Y, Saburi W, Imai R, et al. Evaluation of acceptor selectivity of Lactococcus lactis ssp. lactis trehalose 6-phosphate phosphorylase in the reverse phosphorolysis and synthesis of a new sugar phosphate. Biosci Biotechnol Biochem. 2017;81:1512–1519.
- Tariq MA, Hayashi K. Synthesis of three hetero disaccharides, 4-O-β-glucopyranosyl-6-deoxy-D-glucose, 4-O-β-D-glucopyranosyl-D-mannosamine, and 4-O-β-D-glucopyranosyl-D-mannose, and confirmation of their strucutres by C-13 NMR and MS. Biochem Biophys Res Commun. 1995;214:568–575.
- Hamura K, Saburi W, Abe S, et al. Enzymatic characteristics of cellobiose phosphorylase from Ruminococcus albus NE1 and kinetic mechanism of unusual substrate inhibition in reverse phosphorolysis. Biosci Biotechnol Biochem. 2012;76:812–818.
- Kawahara R, Saburi W, Odaka R, et al. Metabolic mechanism of mannan in a ruminal bacterium, Ruminococcus albus, involving two mannoside phosphorylases and cellobiose 2-epimerase: discovery of a new carbohydrate phosphorylase, β-1,4-mannooligosaccharide phosphorylase. J Biol Chem. 2012;287:42389–42399.
- Sawano T, Saburi W, Hamura K, et al. Characterization of Ruminococcus albus cellodextrin phosphorylase and identification of a key phenylalanine residue for acceptor specificity and affinity to the phosphate group. FEBS J. 2013;280:4463–4473.
- Selinger Z, Schramm M. Enzymatic synthesis of the maltose analogues, glucosyl glucosamine, glucosyl N-acetylglucosamine and glucosyl 2-deoxyglucose by an extract of Neisseria perflava. J Biol Chem. 1961;236:2183–2185.
- Vigsnaes LK, Nakai H, Hemmingsen L, et al. In vitro growth of four individual human gut bacteria on oligosaccharides produced by chemoenzymatic synthesis. Food Funct. 2013;4:784–793.
- Nihira T, Saito Y, Kitaoka M, et al. Identification of Bacillus selenitireducens MLS10 maltose phosphorylase possessing synthetic ability for branched α-d-glucosyl trisaccharides. Carbohydr Res. 2012;360:25–30.
- Inoue Y, Ishii K, Tomita T, et al. Purification and characterization of maltose phosphorylase from Bacillus sp. RK-1. Biosci Biotechnol Biochem. 2001;65:2644–2649.
- Mokhtari A, Blancato VS, Repizo GD, et al. Enterococcus faecalis utilizes maltose by connecting two incompatible metabolic routes via a novel maltose 6´-phosphate phosphatase (MapP). Mol Microbiol. 2013;88:234–253.
- Kamogawa A, Yokobayashi K, Fukui T. Purification and properties of maltose phosphorylase from Lactobacillus brevis. Agric Biol Chem. 1973;37:2813–2819.
- Ehrmann MA, Vogel RF. Maltose metabolism of Lactobacillus sanfranciscensis: cloning and heterologous expression of the key enzymes, maltose phosphorylase and phosphoglucomutase. FEMS Microbiol Lett. 1998;169:81–86.
- Wood BJB, Rainbow C. The maltophosphorylase of beer lactobacilli. Biochem J. 1961;78:204–209.
- Yoshida M, Nakamura N, Horikoshi K. Production and application of maltose phosphorylase and trehalose phosphorylase by a strain of Plesiomonas. Oyo Toshitsu Kagaku. 1995;42:19–25.
- Aisaka K, Masuda T, Chikamune T. Properties of maltose phosphorylase from Propionibacterium freudenreichii. J Ferment Bioengin. 1996;82:171–173.
- Saburi W, Okuyama M, Kumagai Y, et al. Biochemical properties and substrate recognition mechanism of GH31 α-glucosidase from Bacillus sp. AHU 2001 with broad substrate specificity. Biochimie. 2015;108:140–148.
- Ho SN, Hunt HD, Horton RM, et al. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59.
- Mach H, Volkin DB, Burke CJ, et al. Ultraviolet absorption spectroscopy. Methods Mol Biol. 1995;40:91–113.
- Hokse DH. Purification of α‐D‐Glucose‐1‐phosphate. Starch. 1983;35:101–102.
- Miwa I, Okuda J, Maeda K, et al. Mutarotase effect on colorimetric determination of blood glucose with β-D-glucose oxidase. Clin Chim Acta. 1972;37:538–540.
- Cleland WW. The kinetics of enzyme-catalyzed reactions with two or more substrates or products: I. Nomenclature and rate equations. Biochim Biophys Acta. 1963;67:104–137.
- Lowry OH, Lopez JA. The determination of inorganic phosphate in the presence of labile phosphate esters. J Biol Chem. 1946;162:421–428.
- Kelley LA, Mezulis S, Yates CM, et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858.
- Tsumuraya Y, Brewer CF, Hehre EJ. Substrate-induced activation of maltose phosphorylase: interaction with the anomeric hydroxyl group of α-maltose and α-d-glucose controls the enzyme’s glucosyltransferase activity. Arch Biochem Biophys. 1990;281:58–65.