
ABSTRACT
Indoxyl sulfate (IS), a uremic toxin, is a sulfate-conjugated metabolite originated from tryptophan. Accumulating uremic toxins may worsen renal diseases and further complicate related disorders including impaired immune functions under oxidative stress conditions. However, it has remained unclear whether or not IS can directly cause the cellular immune dysfunction. We investigated the effects of IS on the intracellular oxidation level and phagocytic activity in a HL-60-differantiated human macrophage cell model. Incubation of the cells in the presence of IS resulted in increasing intracellular oxidation level and decreasing phagocytic activity. In addition to inhibitors for NADH oxidase (NOX), organic anion transporting polypeptide2B1 (OATP2B1), protein kinase C (PKC), and phosphoinositide 3-kinase (PI3K), a representative antioxidant Trolox, was also shown to significantly relieve the IS-induced oxidation and restore weakened phagocytosis. Collectively, IS may directly down-regulate the phagocytic immune function of macrophages through the oxidation mechanisms including OATP2B1, PKC, PI3K, and NOX pathways.
Abbreviations
CKD: Chronic kidney disease; IS: Indoxyl sulfate; ROS: Reactive oxygen species; NOX: NADH oxidase; OATP2B1: Organic anion transporting polypeptide2B1; PKC: Protein kinase C; PI3K: Phosphoinositide 3-kinase; 2-APT: 2-acetylphenothiazine
Graphical abstract
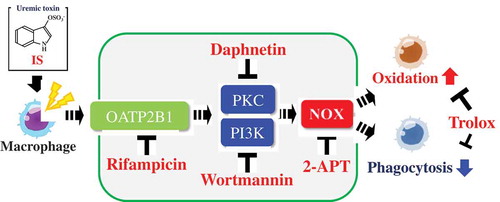
Effects of indoxyl sulfate, a uremic toxin, on the intracellular oxidation level and phagocytic activity using a human macrophage cell model.
Chronic kidney disease (CKD) is known to be associated with increased risk for hemodialysis and cardiovascular diseases. It has been reported that in the United States, 20 million people, more than 10% of the adult population, are at the risk of developing CKD [Citation1], whereas in Japan, 13.3 million people, roughly one in eight in the adult population, are under the same threat [Citation2]. The number of end-stage CKD patients who are on dialysis has been reported to exceed 300,000 in Japan [Citation2,Citation3]. In these patients, uremic toxins may accumulate and exert a range of adverse effects. Uremic toxins may, therefore, cause further renal injury and contribute to the progression of CKD, particularly in patients with end-stage renal disease.
Indoxyl sulfate (IS) is one of the uremic toxins originated from the metabolism of dietary tryptophan [Citation4,Citation5]. In the body, a portion of tryptophan is metabolized to form indole by tryptophanase of intestinal bacteria such as Escherichia coli. Following intestinal absorption, indole may be converted to indoxyl (3-hyrdoxylindole) through Phase I reactions, as mediated by CYP2A6 or CYP2E1, in the liver [Citation6]. Indoxyl may be further metabolized to form sulfate-conjugated IS by hepatic cytosolic sulfotransferase 1A1 (1A1*2) [Citation7] or indoxyl β-glucuronide under the action of UDP-glucurosyltransferase. These potential uremic toxins are usually excreted via the kidney proximal tubules. In patients with renal dysfunction, however, these toxins including IS, may accumulate and remain in circulation. It has been reported that in uremic patients, the level of IS present in blood ranges between 100 to 1,000 µM [Citation8]. It has been suggested that high serum IS level in patients with CKD is associated with the progression of the disease and further complication of related disorders such as impaired immune functions [Citation9,Citation10].
With regard to the progression of renal dysfunction, elevated levels of reactive oxygen species have been suggested as a primary cause [Citation11]. Down-regulated immune system, due to increased oxidative stress, has been observed in uremic patients [Citation9]. Previous reports demonstrated that IS could lead to the generation of reactive oxygen species (ROS) through the activation of cellular NADH oxidase (NOX), in renal proximal tubular cells [Citation12], vascular endothelial cells [Citation13], and smooth muscle cells [Citation14,Citation15]. Increased level of IS promotes monocyte differentiation toward low-inflammatory, profibrotic macrophage cells [Citation16]. However, the mechanism of IS-induced immune dysfunction in uremia is entirely unknown. In this study, we aimed to investigate the effects of IS on the intracellular oxidation level and phagocytic activity in a HL-60-differantiated human macrophage cell model.
Materials and methods
Materials
Dimethyl sulfoxide (DMSO) and rifampicin were products of Nacalai Tesque Inc. (Kyoto, Japan). Indoxyl sulfate (IS), 6-hydroxy-2,5,7,8-tetrametylchromane-2-carboxylic acid (Trolox) were from Sigma Chemicals Co. (Tokyo, Japan). 7,8-Dihydroxycoumarin (daphnetin), 2-acetylphenothiazine (2-APT), 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA), Fluoresbrite fluorescent microspheres (1.0 µm), and wortmannin were purchased from Funakoshi Chemicals Co. (Tokyo, Japan). Phorbol 12-myristate 13-acetate (PMA), RPMI-1640 with L-glutamine, Hank’s balanced salt solution (HBSS) without phenol red, and penicillin-streptomycin solution were from Fujifilm-Wako Pure Chemical Co. (Osaka, Japan). HL-60 human promyelocytic leukemia cells (JCRB00085) were obtained from Japanese Collection of Research Bioresources (Tokyo, Japan). Fetal bovine serum (FBS) was a product of Biowest (Nuaille, France). All other chemicals were of the highest grade commercially available.
Cell culture and preparation of differentiated cells
HL-60 human promyelocytic leukemia cells were routinely maintained under a 5% CO2 atmosphere humidified at 37°C in RPMI-1640 medium supplemented with 5% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin sulfate. To generate the differentiated macrophage cells, HL-60 cells were seeded in a black 96-well plate at a density of 1 × 105 cells/100 µL per well, and incubated for 48 h in the presence of 10 nM PMA as described previously [Citation17]. To prepare the differentiated granulocytic-neutrophil cells, HL-60 cells were seeded in a culture dish at a density of 4 × 105 cells/mL in 10 mL medium and incubated for 6 days in the presence of 1.25% DMSO as described previously [Citation18].
Measurement of intracellular oxidation level
Intracellular ROS formation was evaluated using a probe DCFH-DA [Citation19]. Briefly, the differentiated macrophage cells per well were loaded with 0.1 mM DCFH-DA in HBSS (100 µL) at 37°C for 1 h in a CO2 incubator. The cells were gently washed twice with fresh HBSS (100 µL), and incubated in the presence of IS (10–1,000 µM, 200 µL HBSS per well) at 37°C for 1 h. In the experiments with the inhibitors or an antioxidant Trolox, the cells were preincubated in the presence of individual chemicals in HBSS (200 µL) for 30 min, and then incubated for another 1 h with added IS (plus 10 µL, 1 mM final concentration). The fluorescence intensity (Ex. 480 nm, Em. 530 nm) was measured using a MTP-601 fluorescence microplate reader (Corona Co, Ibaraki, Japan).
The differentiated granulocytic-neutrophil cells prepared in a culture dish or undifferentiated HL-60 cells were suspended in HBSS (10 mL) at 2.5 × 105 cells/mL, and loaded with 0.1 mM DCFH-DA at 37°C for 1 h. The cells were gently washed twice with fresh HBSS (10 mL) in a 15 mL tube, placed in a black 96-well plate (200 µL/well), and incubated in the presence of varying concentrations of IS at 37°C for 1 h. Thereafter, the fluorescence intensity was measured. Trypan blue-unstained viable cells were counted and used in this assay.
Phagocytosis assay
Phagocytosis assay was performed using a previously established method [Citation20]. Briefly, the differentiated macrophage cells prepared in a black 96-well plate were incubated in fresh RPMI medium (200 μL) supplemented with 5% FBS in the presence of varying concentrations of IS and 1% Fluoresbrite fluorescent microspheres for 24 h. In the experiments with the inhibitors or antioxidant Trolox, the cells were incubated in the presence of IS plus individual chemicals in the medium (200 µL) for 24 h. The cells were then gently washed twice with 200 µL fresh HBSS (200 µL). The fluorescence intensity of the fluorescent beads incorporated into the cells (Ex. 365 nm, Em. 492 nm) was measured using a fluorescent microplate reader.
Miscellaneous methods
Cytotoxicity was photometrically evaluated using a commercial Cell Counting Kit-8 (CCK-8) (Dojindo Labs, Kumamoto, Japan). In parallel with the measurement of intracellular oxidation level and phagocytotic activity, cytotoxic effects of IS, inhibitors and Trolox were also determined. No cytotoxic effects of IS, inhibitors or Trolox following a 24-h incubation were detected using CCK-8 (data not shown).
Statistical analysis
For statistical analysis, the values were expressed as mean ± standard deviation based on the data obtained from four independent experiments. Data in part were analyzed using statistical add-on software program (Statcel, OMS Co., Saitama, Japan) for Excel 2016 (Microsoft Co., Redmond, WA, USA). In the Dunnett test, the significant difference was considered at *P< .05 or **P< .01. A post-hoc Tukey–Kramer test was conducted for the multiple comparison and differences at P< .05 were considered significant.
Results and discussion
In patients suffering from renal disorders, the accumulation of uremic toxins including IS (100 to 1,000 µM) [Citation8] is associated with the progression of the disease and further complication of related disorders such as impaired immune functions [Citation9,Citation10]. The current study was designed to investigate the effects of the uremic toxin IS on the intracellular oxidation level and phagocytic activity using an HL-60-differantiated human macrophage cell model.
Elevated intracellular oxidation level upon treatment with IS
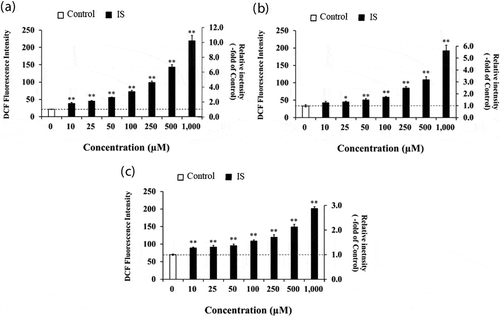
Previous studies have demonstrated that IS may enhance intracellular oxidation in renal proximal tubular cells [Citation12], vascular endothelial cells [Citation13] and smooth muscle cells [Citation14,Citation15]. We first attempted to determine whether IS can modulate intracellular oxidation level in HL-60-differentiated macrophage cells, HL-60-differentiated neutrophil cells and undifferentiated HL-60 cells, using DCFH-DA. As shown in , the results obtained indicated that the intracellular oxidation levels were significantly increased in a concentration-dependent manner upon treatment of macrophages ()), neutrophils ()), and undifferentiated HL-60 cells ()) with IS. The fluorescence intensity of these cells incubated at 1,000 µM IS reached 10.2-fold, 5.74-fold and 2.89-fold, respectively, that of the control. Among the three cell types, IS caused the most prominent enhancement of the oxidation level in macrophages. In a pilot study, we confirmed also that IS could increase the oxidation level in another cell type, human acute monocyte leukemia THP-1 cells.
Figure 1. Effect of IS on the intracellular oxidation level in HL-60-differentiated macrophage cells (a), HL-60-differentiated granulocytic neutrophil cells (b) and undifferentiated HL-60 human leukemic cells (c). DCF fluorescence intensity was fluorometrically determined at Ex. 480 nm and Em. 530 nm. Data shown represent mean ± S.D. from four independent experiments. Using Dunnett test, significant difference from control was considered at *P< .05 or **P < .01. DCF; dichlorofluorescene, IS; indoxyl sulfate.
Suppression of phagocytic activity by IS
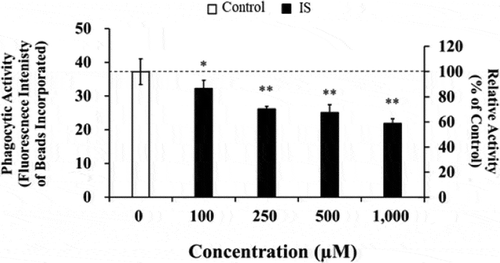
To investigate whether IS is capable of interfering with the immune function of macrophages, we determined the phagocytic activity of the cells incubated in the presence of IS using fluorescent beads. After a 24-h incubation of the cells in the presence of different concentrations of IS, the cellular phagocytic activity was found to decrease gradually and reached 58% at 1,000 µM IS concentration, in comparison with the control (). In contrast, a previous report demonstrated that IS stimulated J774A.1 murine macrophage function and enhanced inflammatory response associated with lipopolysaccharide (LPS), a Gram-negative bacterial endotoxin [Citation21]. Whether the IS can interfere with the inflammatory immune responses, e.g. in LPS-activated human macrophages, still remains to be clarified.
Figure 2. Effect of IS on the phagocytic activity of HL-60-differentiated macrophage cells. Data shown represent mean ± S.D. from four independent experiments. Phagocytic activity was fluorometrically determined at Ex. 365 nm and Em. 492 nm. Using Dunnett test, significant difference from control was considered at *P < .05 or **P < .01. IS; indoxyl sulfate.
Mechanisms underlying the elevated intracellular oxidation level and decreased phagocytic activity upon treatment with IS
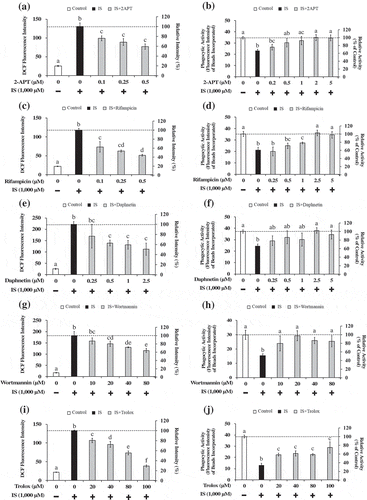
It is an important issue to investigate the possible mechanisms underlying the IS-induced increased intracellular oxidation and decreased phagocytic activity in macrophages. Incubation of the cells in the presence of 2-APT, an inhibitor of NOX, significantly relieved both the elevated cellular oxidation ()) and decreased phagocytic activity ()) as induced by IS. It has been reported that IS could lead to the production of ROS through the activation of NOX in renal proximal tubular cells [Citation12], vascular endothelial cells [Citation13] and smooth muscle cells [Citation14,Citation15]. Moreover, increased ROS production by IS has been also observed in J774A.1 murine monocyte-macrophages [Citation21] and THP-1 human monocytic leukemia cells [Citation16]. Nox family is known to include Nox1, Nox2, Nox3, and Nox4, of which Nox1 and Nox4 are considered the major pro-oxidative enzymes responsible for promoting oxidative stress in the body [Citation22]. Since 2-APT can block Nox1-dependent ROS generation in a concentration-dependent manner [Citation23], it is conceivable that a NOX pathway may be involved in the IS-induced increased oxidation and decreased phagocytic activity in this human macrophage cell model. Likewise, incubation of the cells in the presence of rifampicin, an inhibitor for organic anion transporting polypeptide2B1 (OATP2B1), also significantly ameliorated both the elevated cellular oxidation ()) and decreased phagocytic activity ()), as induced by IS. A previous study demonstrated that IS could promote macrophage activation, thereby inducing vascular inflammation, followed by the uptake of IS through the transporter OATP2B1 [Citation24]. It can be suggested that the organic anion transport via OATP2B1 may be an important pathway for IS to trigger the IS-induced increased oxidation and decreased phagocytosis found in this study. Moreover, incubation of the cells in the presence of daphnetin, an inhibitor of protein kinase C (PKC), significantly relieved both the increased cellular oxidation ()) and decreased phagocytic activity ()), as induced by IS. Similarly, incubation with wortmannin, an inhibitor of phosphoinositide 3-kinase (PI3K), also relieved both the increased cellular oxidation ()) and decreased phagocytic activity ()) caused by IS. PKC and PI3K are the kinases responsible for activating NOX to promote ROS generation [Citation25,Citation26]. Notably, uremic toxin p-cresyl sulfate, a sulfate-conjugated metabolite originated from tyrosine, has been reported to increase ROS generation by activated cellular NOX via the PKC and PI3K pathways in human tubular epithelial cells [Citation27]. Our results seemingly provided support for the involvement of a NOX pathway via the PKC and PI3K in the IS-induced increased oxidation and decreased phagocytosis in our macrophage cell model. We then investigated the effect of Trolox, a representative antioxidant, in the IS-induced immune dysfunction of the human macrophages. Incubation of the cells in the presence of Trolox significantly ameliorated both the increased cellular oxidation ()) and decreased phagocytic activity ()), as induced by IS. Therefore, it appears that antioxidant(s) may be useful agents that can be used to moderate the undesired effects of IS on macrophage immune function. It is an important toxicological and biological issue to clarify whether sulfate conjugation can play a role in the activation or inactivation of indoxyl, a precursor of IS. More works are warranted in order to fully elucidate the mechanisms underlying the IS-induced immune dysfunction.
Organic anion transporters (OATs) and organic anion transporting peptides (OATPs) are known to be involved in the transcellular transport of uremic toxins in the kidney [Citation24]. NOX constitutes a family of pro-oxidative enzymes that function in macrophages and neutrophils to generate the reactive superoxide anion radical for the defense purpose [Citation28]. Additionally, NOX plays an important role in signal transduction in other cell types for cellular stress responses as well as inflammatory responses [Citation29,Citation30]. For instance, activation of NOX in endothelial cells is sensitive to inhibitors of phospholipase A2, PKC, and PI3K [Citation30,Citation31], indicating the involvement of these pathways in NOX activation. Whether phagocytosis and other macrophage-mediated immune responses, e.g. the production of inflammatory cytokines, are involved in the NOX activation pathways awaits further investigation. Macrophage infiltration in the kidney is known as a common feature of CKD [Citation32]. The degree of macrophage infiltration together with the related adverse immune responses correlates with the progression or severity of renal injury. It is noted that infection, bacteremia, and sepsis represent the major threats that underscore the morbidity and mortality in patients with end-stage renal disease [Citation33]. While it remains to be clarified whether or not IS is truly capable of activating macrophage functions as reported [Citation24,Citation34], it is possible that further identification of the chemicals capable of ameliorating the adverse effects of IS in macrophages may provide opportunities for developing strategies for therapeutic intervention for CKD patients.
Figure 3. Effect of 2-APT (an NADH oxidase inhibitor) (a,b), rifampicin (an OATP2B1 inhibitor) (c,d), daphnetin (a PKC inhibitor) (e,f), wortmannin (a PI3K inhibitor) (g,h) and Trolox (an antioxidant) (i,j) on the intracellular oxidation level (a,c,e,g,i) and the phagocytic activity (b,d,f,h,j) of HL-60-differentiated macrophage cells. Data shown represent mean ± S.D. from four independent experiments. Tukey–Kramer’s test was conducted for the multiple comparison and values not sharing a common superscript letter are considered significantly different at P< .05. DCF; dichlorofluorescene, 2-APT; 2-acetylphenothiazine, OATP2B1; organic anion transporter polypeptide2B1, PKC; protein kinase C, PI3K; phosphoinositide 3-kinase, IS; indoxyl sulfate.
In conclusion, we reported for the first time that the uremic toxin IS could directly increase the intracellular oxidation level and decrease the phagocytic activity in the HL-60-differentiated human macrophage cell model. Incubation of the cells with inhibitors of NOX, OATP2B1, PKC, and PI3K, as well as Trolox, was found to help alleviate these adverse effects of IS. Collectively, these results imply that IS may directly down-regulate the phagocytic immune function of macrophages through the oxidation mechanisms including OATP2B1, PKC, PI3K, and NOX pathways. More studies are warranted in order to fully clarify the mechanisms of IS-induced immune dysfunction, thereby help develop strategies for improving the symptoms of uremic patients.
Author contributions
S. Tsutsumi designed the study, mainly carried out experiments, drafted and wrote the manuscript. Y. Tokunaga and S. Shimizu supported performing experiments and data analysis. H. Kinoshita and M. Ono interpreted the data in cellular assays. K. Kurogi, Y. Sakakibara, and M. Suiko interpreted physiological relevance of the sulfated sample. M.-C. Liu interpreted the results and supported English editing to improve the manuscript. S. Yasuda designed the study with S. Tsutsumi, interpreted the results, and wrote the manuscript.
Data availability statement
The data described in this article are openly available in the Open Science Framework at DOI:10.17605/OSF.IO/TPA6U.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Kiefer MM, Ryan MJ. Primary care of the patient with chronic kidney disease. Med Clin North Am. 2015;99:935–952.
- Watanabe H. Molecular mechanisms for uremic toxin-induced oxidative tissue damage via a cardiovascular-renal connection. Yakugaku Zasshi. 2013;133: 889–895. (In Japanese).
- Nitta K, Masakane S, Hanafusa N, et al. 2017 Annual dialysis data report, JSDT renal data registry. Nihon Toseki Igakkai Zasshi. 2018;51: 699–766. (in Japanese).
- Niwa T. Uremic toxicity of indoxyl sulfate. Nagoya J Med Sci. 2010;72:1–11.
- Machado TS, Cerini C, Burtey S. Emerging roles of aryl hydrocarbon receptors in the altered clearance drugs during chronic kidney disease. Toxins (Basel). 2019;11. DOI:10.3390/toxins11040209
- Gillam EMJ, Notley LM, Cai H, et al. Oxidation of indole by cytochrome P450 enzyme. Biochem. 2000;39:13817–13824.
- Banoglu E, King RS. Sulfation of indoxyl by human and rat aryl (phenol) sulfotransferases to form indoxyl sulfate. Eur J Drug Metab Pharmacokinet. 2002;27:135–140.
- Vanholder R, Schepers E, Pletinck A, et al. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: a systematic review. J Am Soc Nephrol. 2014;25:1897–1907.
- Descamps-Latscha B, Jungers P, Witko-Sarsat V. Immune system dysregulation in uremia: role of oxidative stress. Blood Purif. 2002;20:481–484.
- Gao H, Liu S. Role of uremic toxin indoxyl sulfate in the progression of cardiovascular disease. Life Sci. 2017;185:23–29.
- Popolo A, Autore G, Marzocco PS. Oxidative stress in patients with cardiovascular disease and chronic renal failure. Free Radic Res. 2013;47:346–356.
- Shimizu H, Bolati D, Adijiang A, et al. NF-κB plays an important role in indoxyl sulfate-induced cellular senescence, fibrotic gene expression, and inhibition of proliferation in proximal tubular cells. Am J Physiol Cell Physiol. 2011;301:C1201–C1212.
- Masai N, Tatebe J, Yoshino G, et al. Indoxyl sulfate stimulates monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells by inducing oxidative stress through activation of the NADPH oxidase-nuclear factor-κB pathway. Circ J. 2010;74:2216–2224.
- Shimizu H, Hirose Y, Nisijima F, et al. ROS and PDFG-β receptors are critically involved in indoxyl sulfate actions that promote vascular smooth muscle cell proliferation and migration. Am J Physiol Cell Physiol. 2009;297:C389–C396.
- Mutelifu G, Enomoto A, Niwa T. Indoxyl sulfate promotes proliferation of human aortic smooth muscle cells by inducing oxidative stress. J Ren Nutr. 2009;19:29–32.
- Barisione C, Garibaldi S, Furfaro AL, et al. Moderate increase of indoxyl sulfate promotes monocyte transition into profibrotic macrophages. PLoS ONE. 2016;11:e0149276.
- Yasuda S, Kuwata H, Kawamoto K, et al. Effect of highly lipolyzed goat cheese on HL-60 human leukemia cells: antiproliferative activity and induction of apoptotic DNA damage. J Dairy Sci. 2012;95:2248–2260.
- Sugahara S, Ueda Y, Fukuhara K, et al. Antioxidant effects of herbal tea leaves from yacon (Smallanthus sonchifolius) on multiple free radical and reducing power assays, especially on different superoxide anion radical generation systems. J Food Sci. 2015;80:C2420–C2429.
- LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2ʹ,7ʹ-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1992;5:227–231.
- Monobe M, Ema K, Kato F, et al. Technique for screening immune-enhancing polysaccharides in food using 1,25-dihydroxyvitamin D3-differentiated HL60 cells. J Agric Food Chem. 2007;55:2543–2547.
- Adesso S, Popolo A, Bianco G, et al. The uremic toxin indoxyl sulphate enhances macrophage response to LPS. PLoS One. 2013;8:e76778.
- Miyano K, Sumimoto H. Oxidative stress and the NOX family. J Anal Bio-Sci. 2009;32: 289–296. (in Japanese).
- Gianni D, Taulet N, Zhang H, et al. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem Bio. 2010;5:981–993.
- Nakano T, Katsuki S, Chen M, et al. Uremic toxin indoxyl sulfate promotes proinflammatory macrophage activation via the interplay of OATP2B1 and Dll4-Notch signaling. Circulation. 2019;139:78–96.
- Inoguchi T, Sonta T, Tsubouchi H, et al. Protein kinase C-dependent increase in relative oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. J Am Soc Nephrol. 2003;14:S227–S232.
- Yamamori T, Inanami O, Nagahata H, et al. Phosphoinositide 3-kinase regulates the phosphorylation of NADPH oxidase component p47(phox) by controlling cPKC/PKCδ but not Akt. Biochem Biophys Res Commun. 2004;316:720–730.
- Watanabe H, Miyamoto Y, Honda D, et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Inter. 2013;83:582–592.
- Breitenbach M, Rinnerthaler M, Weber M, et al. The defense and signaling role of NADPH oxidases in eukaryotic cells: review. Wien Med Wochenschr. 2018;168:286–299.
- Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair, Pharmacol. Rev. 2011;63:218–242.
- Brandes RP, Weissmann N, Schröder K. Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic Biol Med. 2014;76:208–226.
- O’Donnell RW, Johnson DK, Ziegler LM, et al. Endothelial NADPH oxidase: mechanism of activation by low-density lipoprotein. Endothelium. 2003;10:291–297.
- Guiteras R, Flaquer M, Cruzado JM. Macrophage in chronic kidney disease. Clin Kidney J. 2016;9:765–771.
- Wang HE, Gamboa C, Warnock DG, et al. Chronic kidney disease and risk of death from infection. Am J Nephrol. 2011;34:330–336.
- Wakamatsu T, Yamamoto S, Ito T, et al. Indoxyl sulfate promotes macrophage IL-1β production by activating aryl hydrocarbon receptor/NF-κ/MAPK cascades, but the NLRP3 inflammasome was not activated. Toxins (Basel). 2018;15. DOI:10.3390/toxins10030124