
ABSTRACT
Liver damage induced by ischemia/reperfusion (I/R) remains a primary issue in multiple hepatic surgeries. Innate immune-mediated inflammatory responses during the reperfusion stage aggravate the injury. Nevertheless, the detailed mechanism of hepatic I/R has not been fully clarified yet. Our research focuses on the role of Transducin-like enhancer of split-1 (Tle1) in the liver I/R injury and the relation between Tle1 and Nucleotide-binding oligomerization domain 2 (NOD2). To answer these questions, we constructed mouse models of I/R and cell models of hypoxia/reoxygenation (H/R). We found decreased Tle1 accompanied by increased NOD2 during reperfusion. Mice pro-injected with Tle1-siRNA emerged aggravated liver dysfunction. Repression of Tle1 had a significant impact on NOD2 and downstream NF-κB signaling in vitro. However, alteration of NOD2 failed to affect the expression of Tle1. To conclude, our study demonstrates that Tle1 shelters the liver from I/R injury through suppression of NOD2-dependent NF-κB activation and subsequent inflammatory responses.
Graphical abstract
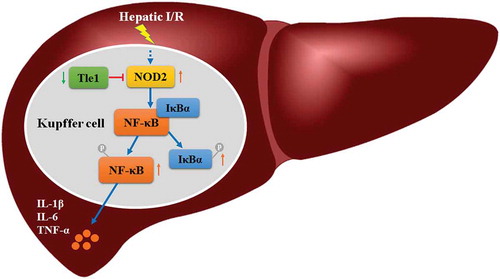
During the hepatic ischemia/reperfusion injury, Tle1 reduces inflammation by regulating the NOD2/NF-κB signaling pathway.
Liver damage induced by ischemia/reperfusion (I/R) remains a noteworthy issue in multiple hepatic pathophysiological processes, such as tumor excision, trauma, and transplantation [Citation1–Citation3]. Liver I/R injury involves both direct cellular damage caused by hypoxia and subsequent aggravating tissue lesions caused by inflammation during the reperfusion stage [Citation4]. Reperfusion contributes to the innate immune-mediated inflammatory response, in which the sentinel pattern recognition receptor (PRR) system in the immune cells senses damage-associated molecular patterns (DAMPs) deriving from necrotic cells. The specialized tissue macrophages residing within the liver sinusoid play an essential role in this process [Citation5]. However, the specific mechanism of liver I/R injury has not been completely identified.
Nucleotide-binding oligomerization domain 2 (NOD2), one member of the NLR family primarily expressed in immunocytes, serves as an intracellular pattern recognition receptor [Citation6]. Once triggered, NOD2-initiating reinforcement of downstream transcription factors gives rise to the generation of inflammatory cytokines, thus being capable of modulating innate immunity and playing an essential part in the development of inflammation [Citation7]. NOD2 has been reported to be prominently upregulated in cerebral microglia and astrocytes during ischemic stroke, and the deletion of NOD2 alleviated the inflammatory responses [Citation8]. Furthermore, activation of NOD2 amplified the myocardial infarction size and accelerated the inflammatory process during cardiac I/R. Conversely, ablation of NOD2 ameliorated the outcomes [Citation9]. However, it remains unclear whether NOD2 is associated with the course of hepatic I/R.
Transducin-like enhancer of split-1 (Tle1), one member of the Groucho family, used to be deemed as a critical corepressor modulating the activities of a large number of critical transcription factors [Citation10]. It can be detected in the brain, liver, and some other organs [Citation11]. Recently, researchers found that Tle1 was more than just a transcriptional corepressor. Results of the assay indicated that NOD2 ignited NF-κB signaling in the inflammatory bowel diseases (IBDs) and Tle1 co-localized with NOD2 in the cytoplasm around the nuclear membrane was capable of abrogating the effects of NOD2 [Citation12]. Hence, we wondered whether Tle1 had an impact on the liver I/R process. Herein, by constructing mouse models of I/R and cell models of hypoxia/regeneration (H/R), we seek to determine the potential function and detailed molecular mechanism of Tle1 in the liver I/R injury, and in doing so, we finally find out Tle1 protects the liver by inhibiting NOD2/NF-κB signaling.
Materials and methods
Animals
Healthy C57BL/6J mice (aged 6 to 8 weeks) were obtained from the Laboratory Animal Center of Chongqing Medical University. All mice were male, weighing 24 to 27 g, brought up under the condition of controlled light, and fed with qualified food and water. All animal protocols were conducted with the approval of the Biomedical Ethics Committee of Chongqing Medical University.
Model of partial hepatic ischemia
Pentobarbital sodium (50 mg per kilogram, intraperitoneal injection) was applied prudently. The mouse hepatic ischemia model was induced based on previous research [Citation13]. After the liver being exposed to ischemia for 1 h, all mice were sacrificed at different timing points (1, 3, 6, 9 h) after reperfusion for collecting tissues and blood samples. Surgical operations performed in the sham-operated groups merely received laparotomy for 1 h without blood occlusion.
In vivo silencing of Tle1 gene
200 nmol/kg scrambled siRNA or Tle1-siRNA (Genepharma, Shanghai, China), mixed with Lipo-fectamine 6000 (Beyotime Biotechnology, Shanghai, China), was injected into mice through the tail vein 12 h before I/R treatment, and mice in the PBS groups were injected with an equal volume of PBS. Samples of serum and tissue were taken from mice after sacrifice.
Aminotransferase assay
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels in the serum were detected using the commercial kits (JianCheng Bioengineering Institute, Nanjing, China) according to the supplier’s standard procedures.
Histopathology
We made use of formalin for fixation after the obtainment of liver tissues. Then liver tissues were paraffin-embedded and cut into sections of 5-μm thickness. Hematoxylin and Eosin (H&E) staining was performed to observe the tissue damage. Suzuki’s score was applied to assess the histopathological changes.
Cell culture
Murine macrophage RAW264.7 cells acquired from the Cell Bank of Chinese Academy of Sciences were cultured in DMEM (Gibco, Grand Island, USA) supplemented with 10% FBS (Gibco, Grand Island, USA) and antibiotics (Beyotime Biotechnology, Shanghai, China) at 37°C in a conventional incubator with 5% carbon dioxide.
Cell hypoxia/reoxygenation model
Cells requiring for H/R treatment were incubated under a hypoxic atmosphere with 1% oxygen, 5% carbon dioxide and 94% nitrogen for 6 h, followed by reoxygenation with 95% air and 5% carbon dioxide for 1, 6, 12 or 24 h, respectively, in terms of previous research [Citation14]. The control groups were incubated in a conventional incubator in the whole process.
In vitro silencing of Tle1 or NOD2 gene
Cells were treated with 50 nM scrambled siRNA, Tle1 siRNA, or NOD2 siRNA transfection, respectively, for 24 h prior to H/R treatment, using Lipofectamine 6000 (Beyotime Biotechnology, Shanghai, China) according to the manufacturers’ manuals. After transfection, cells were subjected to H/R treatment.
Cytokine measurement
ELISA kits bought from Neobioscience (Neobioscience, Beijing, China) were used to measure the inflammatory cytokines (IL-6, TNF-α, and IL-1β) in cell supernatant or mouse serum.
qRT-PCR assay
Extraction of total RNA from the hepatic tissues or RAW264.7 cells was conducted through the usage of Trizol (Beyotime Biotechnology, Shanghai, China). The cDNA Synthesis SuperMix (Bimake, Houston, TX, USA) was applied to convert the total RNA to cDNA. Then, Quantitative real-time polymerase chain reaction amplification using 2x SYBR Green qPCR Master Mix (Bimake, Houston, TX, USA) was performed to quantify the expression of mRNA. Each experiment was performed in triplicate, and β-actin acts as a reference gene. Sequences for synthesizing primers are listed below: Tle1 forward, 5′-CCA GTA CCT CTC ACG CCT CA-3′, and reverse, 5′-GCC CAC TCA GAG CAC TAG AC-3′; NOD2 forward, 5′-CAG AAA CTA GCT CTC TTC AAC AAC A-3′, and reverse, 5′-GTG ATT GTT CCC CAC CCT CA-3′; TNF-α forward, 5′-CAT CTT CTC AAA ATT CGA GTG ACA A-3′, and reverse, 5′-TGG GAG TAG ACA AGG TAC AAC CC-3′; β-actin forward, 5ʹ-GGC TGT ATT CCC CTC CAT CG-3ʹ, and reverse, 5ʹ-CCA GTT GGT AAC AAT GCC ATG T-3ʹ.
Western blot assay
RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) added with phosphatase inhibitor (Beyotime Biotechnology, Shanghai, China) and protease inhibitor cocktail (Beyotime Biotechnology, Shanghai, China) was used for lysis of cultured cells. Protein levels were detected by employing the BCA Protein Assay Kit (Beyotime Biotechnology, Shanghai, China). Then samples were electrophoresed on SDS-PAGE and transferred onto PVDF membranes. After being blocked with the Blocking Buffer for Western Blot (Beyotime Biotechnology, Shanghai, China), the membranes were incubated with corresponding primary antibodies at 4 degrees Celsius overnight, followed by incubation with appropriate secondary antibodies the next day. Images were visualized by using the ECL Chemiluminescence (Advansta, CA, USA). Primary antibodies for Western blot are as follows: Tle1 (Abcam, Cambridge, UK), p65, p-p65 (Proteintech, Rosemont, USA), NOD2, p-IκBα, IκBα (Santa Cruz Biotechnology, Santa Cruz, USA), IL-1β (Wanleibio, Shanghai, China), β-actin (Beyotime Biotechnology, Shanghai, China).
Statistical analysis
We made use of the SPSS Statistics 22.0 to carry out statistical analysis. All results were obtained from at least three independent repeats. Student’s t-test was applied to assess statistical significance between two groups. Differences among groups were tested by one-way ANOVA. A p-value below 0.05 was considered statistically significant.
Results
Decreased Tle1 accompanied by increased NOD2 is detected during mouse hepatic I/R process
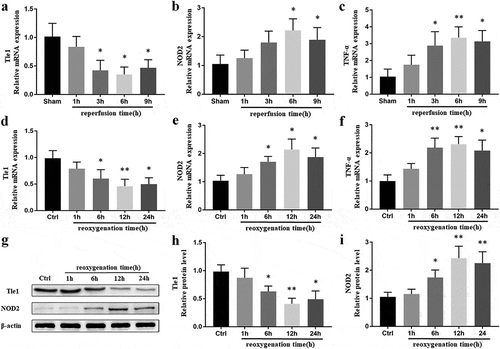
To analyze the expression of Tle1 and NOD2 in the liver tissues during I/R injury, mouse models of partial liver ischemia were established. Compared with the Sham groups, I/R treatment led to a substantial decrease of Tle1 in mRNA levels, reaching a minimum at 6 h after reperfusion ()), but NOD2 was significantly enhanced in the meantime ()). Inflammatory cytokine TNF-α was increased as well, indicating the progression of inflammatory process during I/R ()). To verify the expression of Tle1 and NOD2 in vitro after H/R treatment, we constructed cell models of H/R. Similar to the results achieved in mouse models of liver I/R injury, dramatically elevated NOD2 and largely depressed Tle1 in comparison to the control groups in both mRNA and protein levels were observed (–i)). At last, we identified 6 h of hypoxia, followed by 12 h of reoxygenation as conditions for further experiments. These results suggest that Tle1 and NOD2 might be connected during liver I/R injury.
Figure 1. Expression of Tle1, NOD2, and TNF-α in the liver I/R model both in vivo and in vitro.
(a-c) After mice treated with ischemia for 1 h, followed by reperfusion for 1, 3, 6, 9 h, the mRNA levels of NOD2, Tle1, and TNF-α were detected. (d-i) After hypoxia for 6 h and subsequent reoxygenation for 1, 6, 12, 24 h, the mRNA and protein levels of Tle1 and NOD2 as well as the mRNA levels of TNF-α in the cells were measured. *p < 0.05 and **p < 0.01 in comparison to the Sham or Ctrl groups.
Tle1 inhibits I/R injury and prevents the release of pro-inflammatory cytokines
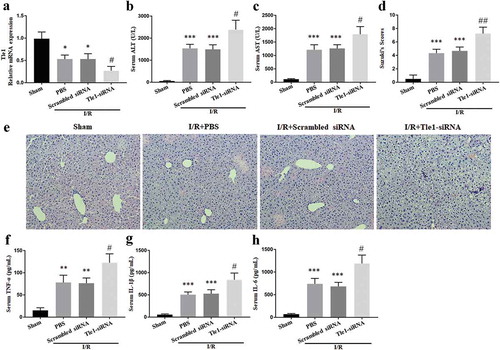
To reveal the in vivo function of Tle1 in the liver I/R injury, mice were injected with Tle1-siRNA from the tail vein before surgical operations. Mouse livers pretreated with Tle1-siRNA led to a considerable drop in Tle1 expression ()). The serum levels of aminotransferase (ALT and AST) were elevated in groups subjected to I/R treatment, whilst there was a more marked elevation in the Tle1-siRNA groups, suggesting severer hepatic damage (-c)). Further, to assess the severity of liver damage, HE staining was applied to observe pathological changes of the liver tissues. Compared to the scrambled siRNA groups, knockdown of Tle1 increased the histopathological alterations (-e)). In the meantime, serum pro-inflammatory agents such as IL-6, TNF-α, and IL-1β were elevated in the Tle1-siRNA groups (-h)). Results in this chapter show that the abolishment of Tle1 aggravates the inflammation and tissue damage during liver I/R, thus indicating the protective role of Tle1.
Figure 2. Effects of Tle1 in vivo. Mice were subjected to ischemia for 1 h, followed by reperfusion for 6 h.
(a) The mRNA levels of Tle1 in mice treated with PBS, scrambled siRNA, or Tle1-siRNA. (b-c) Serum concentrations of ALT and AST. (d-e) H&E staining of liver tissues (200× magnification) and Suzuki’s scores. (f-h) Serum concentrations of inflammatory agents (IL-6, TNF-α, and IL-1β). *p < 0.05, **p < 0.01 and ***p < 0.001 in comparison to the Sham groups. #p < 0.05 and ##p < 0.01 in comparison to the Scrambled siRNA groups.
Tle1 suppresses the expression of NOD2/NF-κB signaling pathway-related molecules
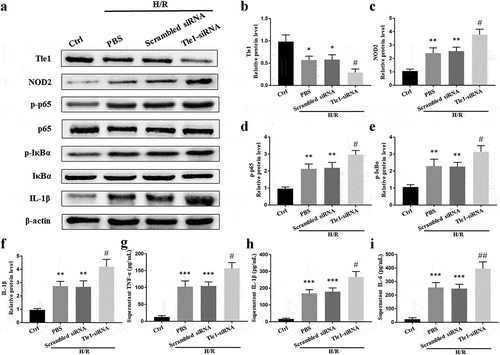
To assess the expression of NOD2 and activation of NF-κB signaling in vitro during H/R when Tle1 was down-regulated, cells were transfected with Tle1-siRNA. Western blot analysis showed that compared with the scrambled siRNA groups, protein expression of NOD2 in the Tle1-siRNA groups was increased and the NF-κB signaling was strongly ignited as demonstrated by phosphorylation of p65, phosphorylation of IκBα as well as elevation of IL-1β (-f)). Besides, the downregulation of Tle1 resulted in increased release of inflammatory agents (IL-6, TNF-α, and IL-1β) in cell supernatant (-i)). Overall, we conclude that Tle1 is able to repress the NOD2/NF-κB signaling.
Figure 3. Effects of Tle1 on NOD2 and NF-κB signaling in vitro.
(a-f) The protein levels of Tle1, NOD2, p-p65, p65, p-IκBα, IκBα, and IL-1β. (g-i) Inflammatory agents (IL-6, TNF-α, and IL-1β) in cell supernatant. *p < 0.05, **p < 0.01 and ***p < 0.001 in comparison to the Ctrl groups. #p < 0.05 and ##p < 0.01 in comparison to the Scrambled siRNA groups.
Tle1 inhibits NF-κB signaling by regulating NOD2
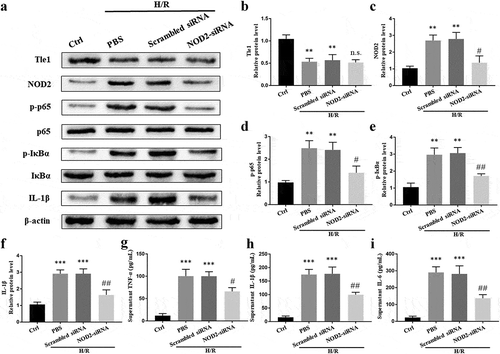
To further clarify the relation between Tle1 and NOD2, cells were treated with NOD2-siRNA. Compared with the scrambled siRNA groups, the expression of NF-κB pathway-related proteins was significantly decreased in NOD2-siRNA groups (-f)). Moreover, downregulation of NOD2 led to the reduced release of IL-6, TNF-α, and IL-1β in cell supernatant (-i)). Intriguingly, inhibition of NOD2 failed to affect the expression of Tle1 (-b)).
Figure 4. Effects of NOD2 on Tle1 and NF-κB signaling in vitro.
(a-f) The protein levels of Tle1, NOD2, p-p65, p65, p-IκBα, IκBα, and IL-1β. (g-i) Serum levels of IL-6, TNF-α, and IL-1β. **p < 0.01 and ***p < 0.001 in comparison to the Ctrl groups. n.s. p > 0.05, #p < 0.05 and ##p < 0.01 in comparison to the Scrambled siRNA groups.
Discussion
In our current study, we made an inquiry into whether Tle1 made a difference in the liver I/R process. Our data indicate that Tle1 relieves the inflammatory responses during liver I/R injury via regulating the NOD2/NF-κB signaling.
Tle1 is expressed in a variety of normal tissues and cancer tissues, especially in synovial sarcoma, where it acts as a biological marker for diagnosis [Citation15]. Notably, Tle1 located in human macrophages has been proved to present a prominent anti–inflammatory effect [Citation11]. Furthermore, when mouse macrophages challenged with LPS, Tle1 has the capacity to relieve the inflammation [Citation16]. In our study, we detected a marked decrease of Tle1 in mouse models of I/R and cell models of H/R, which may imply that Tle1 played a vital role during liver I/R injury. Besides, knockdown of Tle1 in mice caused severer liver damage, as was indicated by elevated serum transaminase (ALT and AST) levels and deteriorated hepatic histological alterations. Thus, we find that Tle1 is an essential modulator able to shelter the liver from I/R injury.
The previous study has addressed the crucial role of NOD2 in promoting inflammatory responses in concanavalin A (ConA)-induced hepatitis [Citation17]. Additionally, NOD2 was reported to take part in the heart, brain, and intestine I/R injury [Citation8,Citation9,Citation18–Citation20]. These studies illustrated that NOD2 might promote the inflammation in the liver I/R injury. Recently, Tle1 was shown to be capable of suppressing the inflammation deriving from NOD2 activation [Citation12]. Thus, we suspected that Tle1 could reduce the inflammation during liver I/R injury through the suppression of NOD2 signaling pathway. To verify our speculations, cells were transfected with Tle1-siRNA before H/R treatment in the first place. Considering that NOD2 modulates diverse downstream signaling pathways, among which NF-κB is recognized as a key signaling pathway controlling the synthesis of pro-inflammatory agents in liver I/R injury [Citation21–Citation25], we simultaneously investigated the expression of NOD2 and NF-κB signaling pathway-related molecules, including p-p65 and p-IκBα. We found a significantly upregulated expression of these proteins after H/R, which indicated the ignition of NF-κB signaling. Moreover, the downregulation of Tle1 gave rise to the increased release of inflammatory cytokines. We can conclude from these results that Tle1 is able to regulate NOD2 and NF-κB signaling pathway. Afterward, to better understand the relation between Tle1 and NOD2, cells were transfected with NOD2 siRNA before H/R treatment. It was worth noting that the downregulation of NOD2 failed to affect the expression of Tle1. Therefore, based on the above, we infer that Tle1 is the suppressor of NOD2 during H/R.
Tle1 used to be deemed as a corepressor interacting with diverse DNA-binding transcription factors. Tle1 represses NF-κB activity under certain circumstances [Citation16]. In our study, we focus on the relation between Tle1 and NOD2 in the liver I/R injury, and the role of Tle1 serving as a corepressor of NF-κB under the circumstance of I/R requires further confirmation. NOD2 is known to bind the Receptor Interacting Serine/Threonine Kinase 2 (RIPK2), triggering activation of NF-kB and MAPKs signaling pathway [Citation26]. A growing body of evidence has verified the significance of MAPKs signaling in the I/R-induced liver damage [Citation27–Citation29]. Nevertheless, this study merely focuses on NF-κB regardless of MAPKs, which is supposed to be assessed in a future study.
In summary, our study demonstrates that Tle1 possesses a protective function on the liver I/R injury. The detailed mechanisms rely on the suppression of pattern recognition receptor NOD2, resulting in depressed activation of NF-κB signaling followed by subsequent restraint of the inflammation progression. Thus, we propose Tle1 can serve as a latent target to ameliorate liver I/R injury.
Author contributions
Zhongjun Wu and Wei Chen designed the experiments. Tong Mou, Daofeng Zheng, Junliang Pu, Jiangwen Dai, and Wei Chen carried out the experiments and performed the statistical analysis. Zuotian Huang, Yunhai Luo, and Yuke Zhang drafted the manuscript. All authors read and approved this paper.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Peralta C, Jimenez-Castro MB, Gracia-Sancho J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J Hepatol. 2013;59(5):1094–1106.
- Gracia-Sancho J, Casillas-Ramirez A, Peralta C. Molecular pathways in protecting the liver from ischaemia/reperfusion injury: a 2015 update. Clin sci. 2015;129(4):345–362.
- Jia JJ, Li JH, Jiang L, et al. Liver protection strategies in liver transplantation. Hbpd Int. 2015;14(1):34–42.
- Qin JJ, Mao W, Wang X, et al. Caspase recruitment domain 6 protects against hepatic ischemia/reperfusion injury by suppressing ASK1. J Hepatol. 2018;69(5):1110–1122.
- Zhai Y, Petrowsky H, Hong JC, et al. Ischaemia-reperfusion injury in liver transplantation–from bench to bedside. Nat Clin Pract Gastroenterol Hepatol. 2013;10(2):79–89.
- Normand S, Waldschmitt N, Neerincx A, et al. Proteasomal degradation of NOD2 by NLRP12 in monocytes promotes bacterial tolerance and colonization by enteropathogens. Nat Commun. 2018;9(1):5338.
- Ting JP, Duncan JA, Lei Y. How the noninflammasome NLRs function in the innate immune system. Science (New York, NY). 2010;327(5963):286–290.
- Liu H, Wei X, Kong L, et al. NOD2 is involved in the inflammatory response after cerebral ischemia-reperfusion injury and triggers NADPH oxidase 2-derived reactive oxygen species. Int J Biol Sci. 2015;11(5):525–535.
- Liu Y, Yang H, Liu LX, et al. NOD2 contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and inflammation. Life Sci. 2016;149:10–17.
- Jennings BH, Ish-Horowicz D. The Groucho/TLE/Grg family of transcriptional co-repressors. Genome Biol. 2008;9(1):205.
- De Paoli F, Copin C, Vanhoutte J, et al. Transducin-like enhancer of split-1 is expressed and functional in human macrophages. FEBS Lett. 2016;590(1):43–52.
- Nimmo ER, Stevens C, Phillips AM, et al. TLE1 modifies the effects of NOD2 in the pathogenesis of Crohn’s disease. Gastroenterology. 2011;141(3):972–981.e1-2.
- Abe Y, Hines IN, Zibari G, et al. Mouse model of liver ischemia and reperfusion injury: method for studying reactive oxygen and nitrogen metabolites in vivo. Free Radic Biol Med. 2009;46(1):1–7.
- Wang X, Mao W, Fang C, et al. Dusp14 protects against hepatic ischaemia-reperfusion injury via Tak1 suppression. J Hepatol. 2017;68(1):118–129.
- Terry J, Saito T, Subramanian S, et al. TLE1 as a diagnostic immunohistochemical marker for synovial sarcoma emerging from gene expression profiling studies. Am J Surg Pathol. 2007;31(2):240–246.
- Ramasamy S, Saez B, Mukhopadhyay S, et al. Tle1 tumor suppressor negatively regulates inflammation in vivo and modulates NF-kappaB inflammatory pathway. Proc Natl Acad Sci U S A. 2016;113(7):1871–1876.
- Body-Malapel M, Dharancy S, Berrebi D, et al. NOD2: a potential target for regulating liver injury. Lab Invest. 2008;88(3):318–327.
- Zhang H, Zhu T, Liu W, et al. TIPE2 acts as a negative regulator linking NOD2 and inflammatory responses in myocardial ischemia/reperfusion injury. J Mol Med (Berl). 2015;93(9):1033–1043.
- Chen L, Kong L, Wei X, et al. beta-arrestin 2 negatively regulates NOD2 signalling pathway through association with TRAF6 in microglia after cerebral ischaemia/reperfusion injury. J Cell Mol Med. 2019;23(5):3325–3335.
- Perez-Chanona E, Muhlbauer M, Jobin C. The microbiota protects against ischemia/reperfusion-induced intestinal injury through nucleotide-binding oligomerization domain-containing protein 2 (NOD2) signaling. Am J Pathol. 2014;184(11):2965–2975.
- Mukherjee T, Hovingh ES, Foerster EG, et al. NOD1 and NOD2 in inflammation, immunity and disease. Arch Biochem Biophys. 2019;670:69–81.
- Kersse K, Bertrand MJ, Lamkanfi M, et al. NOD-like receptors and the innate immune system: coping with danger, damage and death. Cytokine Growth Factor Rev. 2011;22(5–6):257–276.
- Platnich JM, Muruve DA. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys. 2019;670:4–14.
- Li Z, Zhang J, Mulholland M, et al. mTOR activation protects liver from ischemia/reperfusion-induced injury through NF-kappaB pathway. Faseb J. 2017;31(7):3018–3026.
- Shin T, Kuboki S, Lentsch AB. Roles of nuclear factor-kappaB in postischemic liver. Hepatol Res. 2008;38(5):429–440.
- Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27(4):549–559.
- Uehara T, Bennett B, Sakata ST, et al. JNK mediates hepatic ischemia reperfusion injury. J Hepatol. 2005;42(6):850–859.
- Yan ZZ, Huang YP, Wang X, et al. Integrated omics reveals tollip as an regulator and therapeutic target for hepatic ischemia-reperfusion injury in mice. Hepatology. 2019;70(5):1750–1769.
- Jimenez-Castro MB, Cornide-Petronio ME, Gracia-Sancho J, et al. Mitogen activated protein kinases in steatotic and non-steatotic livers submitted to ischemia-reperfusion. Int J Mol Sci. 2019;20:7.