
ABSTRACT
MiR-181a-5p’s mechanism in hypoxia–reoxygenation (H/R)-induced cardiomyocytes apoptosis has not been clarified. This study verified that SIRT1 was the target of miR-181a-5p. MiR-181a-5p expression was up-regulated or down-regulated in H/R-induced cardiomyocytes, and SIRT1 was transfected into cells alone or in combination with miR-181a-5p. Cell viability, apoptosis, levels of released lactate dehydrogenase (LDH), malondialdehyde (MDA), and superoxide dismutase (SOD), as well as the Bcl-2, Bax, and Caspase 3 levels in treated cells were tested. On the one hand, down-regulated miR-181a-5p promoted cell viability, reduced released LDH and MDA, and increased SOD level in H/R-induced cardiomyocytes. On the other hand, miR-181a-5p inhibited apoptosis and elevated Bcl-2 expression while decreasing the expressions of Bax and Caspase 3 in treated cells, but the effects of miR-181a-5p could be rescued by SIRT1. In conclusion, miR-181a-5p involved in H/R-induced cardiomyocytes apoptosis through regulating SIRT1, which might become a novel direction for related diseases.
GRAPHICAL ABSTRACT
SIRT1 counteracted the effects of over-expressed miR-181a-5p on hypoxia/reoxygenation (H/R)-induced cardiomyocytes.
Abbreviations
H/R: hypoxia–reoxygenation; qRT-PCR: quantitative reverse transcription-polymerase chain reaction; WB: western blot; MDA: malondialdehyde; LDH: lactate dehydrogenase; SOD: superoxide dismutase; AMI: Acute myocardial infarction; I/R: ischemia–reperfusion; DNA: Deoxyribonucleic acid; miRNAs: microRNA; mRNA: messenger RNAs; 3ʹ UTR: 3ʹ-untranslated region; NC: negative control; OD: optical density; PI: propidium iodide; PVDF: polyvinylidene fluoride
Acute myocardial infarction (AMI) is the primary element of death in patients with cardiovascular disease, and timely opening of infarct-related arteries and restoration of blood supply to ischemic myocardium is the key to rescue dying myocardial cells [Citation1]. However, the resumption of blood perfusion after a period of time of myocardial ischemia will lead to aggravation of myocardial structural and functional damage, thereby increasing the incidence of myocardial depression, decreasing cardiac function and malignant arrhythmia, and such a process is known as myocardial ischemia–reperfusion (I/R) injury [Citation2–Citation4]. Studies showed that cardiomyocytes apoptosis, which is a main characteristic change of myocardial I/R injury, could determine the severity and plays a pivotal role in the prognosis of patients [Citation5–Citation7]. Moreover, oxidative stress occurred in the process of I/R injury will increase damages to cell desoxyribonucleic acid (DNA) and membranes, ultimately leading to the death of cardiomyocytes [Citation8,Citation9]. Hypoxia/reoxygenation (H/R) in cells is a main characteristic of I/R and is frequently used to simulate the activity of I/R [Citation10,Citation11]. Low cell viability and high apoptosis inflammation response appear after cell injury induced by H/R [Citation12]. Jennings et al. first confirmed myocardial ischemia–reperfusion injury and observed that reperfusion accelerates the progression of cell necrosis [Citation13]. Myocardial ischemia–reperfusion injury is one of the leading causes of myocardial infarction [Citation13]. In this study, we sought to understand the underlying molecular mechanisms of myocardial ischemia–reperfusion injury in order to find treatment strategies to improve acute myocardial infarction.
MicroRNAs (miRNAs) are RNAs with short sequences (with about 18 to 25 nucleotides in length), and due to they lack open-reading frame, miRNAs do not have protein-encoding potential [Citation14]. MiRNAs can combine with messenger RNAs (mRNA) transcribed from specific genes to block mRNA translation or directly degrade it, and finally leading to the silence of target genes [Citation15]. Recently, studying the regulatory role of miRNAs in cardiac I/R injury has attracted much research attention, and multiple miRNAs have been confirmed to have important effects on the course of myocardial I/R injury [Citation16]. For example, Qin D et al. [Citation17] showed that miR-223-5p and miR-223-3p could synergistically alleviate cardiac necroptosis stimulated by I/R injury; Diao H et al. [Citation18] demonstrated that miR-210 relieved cardiomyocyte apoptosis caused by oxidative stress. By reviewing previous studies, we found that abnormally expressed miR-181a is closely related to the clinical prognosis of various human cancers, and it exerts a momentous function in regulating cell apoptosis [Citation19,Citation20].
In the current research, hypoxia/reoxygenation (H/R) models of rat cardiomyocytes H9C2 were structured to establish I/R injury, and the expression of miR-181a-5p in cardiomyocytes was up-regulated and down-regulated. This study also analyzed the effects of miR-181a-5p on cell viability, apoptosis, and oxidative stress-related indicators, and further detected its potential target and mechanism. The objective of our report was to explore the functions of miR-181a-5p in cardiomyocytes apoptosis, hoping to determine underlying therapeutic targets for treating I/R injury-related diseases.
Material and methods
Cell culture
Rat H9C2 cardiomyocytes were supplied by the Cell Center of Chinese Academy of Sciences (Shanghai, China), and cultured in Dulbecco’s modified Eagle medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, Carlsbad, CA, USA) and 1% penicillin/streptomycin (Gibco, USA). The cells were maintained in a humidified condition at 37°C with 5% CO2.
Luciferase reporter assay
Targetscan 7.2 (http://www.targetscan.org/.) showed that the position 67–73 of SIRT1 3ʹ UTR could bind to miR-181a-5p, and the prediction was further verified by performing luciferase reporter assay. For the assay, the mutant (MUT) or wild type (WT) SIRT1 3ʹ UTR sequence was installed into the reporter plasmid (Promega, Madison, WI, USA), and respectively co-transfected into H9C2 cells with or without miR-181a-5p using LipofectamineTM 2000 transfection reagent (Invitrogen, Carlsbad, California, USA). After incubation for 48 h, the cells were collected for the measurement of luciferase viability using Luciferase Assay Systems (Promega, Madison, Wisconsin, USA).
Cell treatment and transfection
MiR-181a-5p mimic, inhibitor, and negative control (NC) were severally transfected into untreated H9C2 cells for the measurement of miR-181a-5p expression. Moreover, H9C2 cells were maintained in by hypoxia (85% N2, 10% H2, 5% CO2, and <0.1% O2) for 24 h, and followed by reoxygenation for 24 h to trigger H/R injure in a humidified atmosphere with 5% CO2 and 95% air at 37°C [Citation21]. Next, miR-181a-5p mimic and antagonist were transfected into H/R-induced H9C2 cells alone or in combination, and the cells transfected with scramble sequence served as NC. In addition, the SIRT1 sequence was synthesized by RiboBio and cloned into the pcDNA3.1 vector (60908–1440, Tiandz, Beijing, China) and combined with NC or mimic were transfected into H9C2 cells. Transfection experiments were implemented by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer instructions. All plasmids and vectors used in this study were constructed by GenePharma Co., Ltd. (Shanghai, China).
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
The expression levels of miR-181a-5p and SIRT1 in H/R-induced H9C2 cells were determined by qRT-PCR assays, with GAPDH and U6 served as the internal references. Total RNAs of cells were isolated using TRIzol reagent (Invitrogen, Carlsbad, California, USA), and the quality and integrity of isolated RNA were determined by spectrophotometer (NanoDrop-2000c; Thermo Fisher Scientific, Massachusetts, USA) and 1% agarose-modified gel electrophoresis. One microgram of the total RNA was reverse-transcribed into cDNA by the PrimeScript RT Master Mix Perfect Real-Time (TaKaRa, Shiga, Japan) following the manufacturer’s instructions. qRT-PCR assay was performed by the ABI Prism 7500 Fast Real-time PCR System (Applied Biosystems, Foster City, CA) under the following conditions: pre-denaturation at 95°C for 10 min and then 40 cycles at 95°C for 10 s, and at 60°C for 60 s. The sequences of primers were synthesized by Gene Pharma (Shanghai, China) and are shown in . The data of relative mRNA levels were analyzed by the comparative 2−ΔΔCt method [Citation22].
Table 1. Primer base sequence.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
The viability of H/R-induced H9C2 cells after cell transfection was measured by MTT assay. Briefly, 48 h after the transfection, 0.5 mg/mL of MTT reagent (Sigma-Aldrich, USA) was added into the cells at 37°C to incubate for another 4 h. The optical density (OD) of cells was read using the ELX-800 Biotek plate reader (Winooski, USA) at 570 nm.
Cell apoptosis
The apoptosis rate of H/R-induced H9C2 cells after the transfection was determined by a flow cytometry. In brief, H/R-induced H9C2 cells after cell transfection were collected and stained by 10 μL Annexin V and 5 μL propidium iodide (PI), and the proportion of cell apoptosis was measured by a FACSCalibur flow cytometer (BD Biosciences, USA) according to the manufacturer’s instructions.
Indicators analysis
The levels of lactate dehydrogenase (LDH), malondialdehyde (MDA), and superoxide dismutase (SOD) of H/R-induced H9C2 cells after cell transfection were determined using the corresponding kits (Nanjing Jiancheng Bioengineering Institute, China) by colorimetric methods. The absorbance was measured by an automatic enzyme standard instrument (STAT FAX 2100, Awareness Technology, Inc. USA). All operations were performed in accordance with the manufacturer’s instructions.
Western blotting (WB) analysis
The expressions of proteins derived from H/R-induced H9C2 cells were determined by WB. The total proteins of the cells were extracted by RIPA lysis buffer (Beyotime, Shanghai, China), and the protein concentration was determined by Bicinchoninic Protein Assay kit (BCA, Pierce, Rockford, IL, USA). Fifty micrograms of extracted protein was isolated on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, Beyotime, Shanghai, China) and then transferred onto polyvinylidene fluoride (PVDF) membranes. Subsequently, the membranes were sealed by 5% nonfat milk for 2 h and incubated with the primary antibodies SIRT1 (0.5 µg/mL, ab110304, Abcam, USA), Bcl-2 (1:1000, ab59348, Abcam, USA), Bax (1:2000, ab32503, Abcam, USA), Caspase 3 (1 µg/mL, ab2302, Abcam, USA) overnight at 4°C, with GAPDH (1:1000, ab8245, Abcam, USA) served as the internal reference. The corresponding secondary antibodies goat anti-mouse IgG H&L (HRP; 1:1000, ab150113, Abcam) and goat anti-rabbit IgG H&L (HRP; 1:7000, ab97051, Abcam, USA) were added at room temperature and incubated for another 1 h. The band signals were developed by the Enhanced Chemiluminescence Western Blot Detection Kits (Thermo Fisher, MA, USA).
Statistical analysis
Statistical Package of the Social Sciences 20.0 software (SPSS, Inc., Chicago, USA) was used for data analysis. The data were shown as mean ± standard deviation (SD). The comparison between two groups was performed by Student’s t-test, and One-way ANOVA with Dunnett’s post hoc test was implemented for multiple group analysis. All experiments were performed in triplicate. P < 0.05 was considered to be statistically significant.
Results
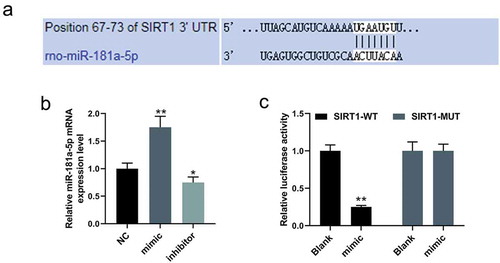
SIRT1 was the target gene of miR-181a-5p
Targetscan 7.2 predicted that the position 67–73 of SIRT1 3ʹ UTR was the targeted area of miR-181a-5p ()). The experimental results of qRT-PCR assays indicated that the expression of miR-181a-5p in rat cardiomyocytes was successfully elevated by mimic, and effectively suppressed by the inhibitor (P < 0.05, )). Moreover, in luciferase reporter assay, the luciferase viability was notably decreasing in cardiomyocytes transfected with SIRT1-WT plus miR-181a-5p mimic (P < 0.001, )).
Figure 1. SIRT1 was the target gene of miR-181a-5p.
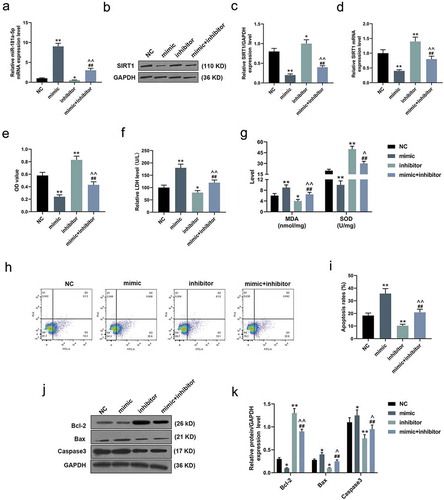
Up-regulated miR-181a-5p suppressed the viability of H/R-induced cardiomyocytes and accelerated cell apoptosis
As shown in ), qRT-PCR assay revealed that miR-181a-5p expression level in H/R-induced cardiomyocytes was significantly increased after transfection with mimic, but decreased under the action of inhibitor; however, co-transfection of these two could neutralize the effects of the mimic and inhibitor (P < 0.05). In addition, both qRT-PCR and WB experiments demonstrated that the expression of SIRT1 in H/R-induced cardiomyocytes was effectively suppressed by over-expressed miR-181a-5p, but promoted by knockdown miR-181a-5p (P < 0.05, )). MTT experiments showed that over-expressed miR-181a-5p reduced the viability of H/R-induced cardiomyocytes, while down-regulated miR-181a-5p directly increased the cell viability and reversed the anti-cellular viability of up-regulated miR-181a-5p (P < 0.001, )). Furthermore, we also found that up-regulated miR-181a-5p increased LDH release and MDA levels but reduced SOD level in H/R-induced cardiomyocytes, which was completely opposite to the effects of down-regulated miR-181a-5p (P < 0.05, ,g)). Furthermore, according to flow cytometry analysis, the over-expression of miR-181a-5p elevated the apoptosis rate of H/R-induced cardiomyocytes, while its inhibition noticeably suppressed cell apoptosis (P < 0.001, ,i)). Accordingly, in the detection of apoptosis-related proteins, WB assay revealed that up-regulated miR-181a-5p inhibited the expressions of Bcl-2 and promoted Bax and Caspase 3 expression in H/R-induced cardiomyocytes (P < 0.05, ,k)).
Figure 2. Up-regulated miR-181a-5p suppressed the viability of hypoxia/reoxygenation (H/R)-induced cardiomyocytes and accelerated cell apoptosis.
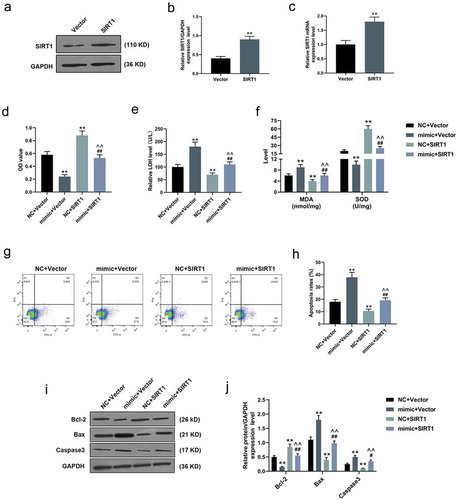
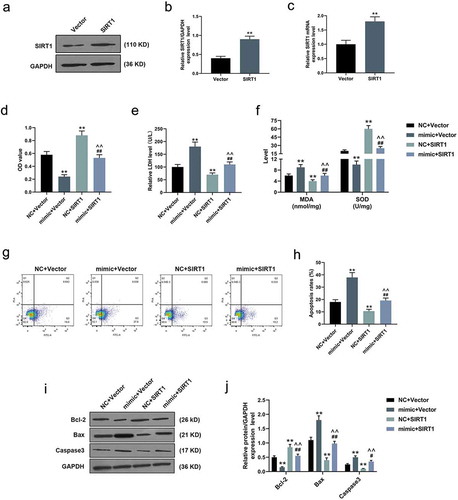
SIRT1 counteracted the effects of over-expressed miR-181a-5p on H/R-induced cardiomyocytes
In the following transfection experiments, both qRT-PCR and WB experiments confirmed the obvious transfection effect of SIRT1 in H/R-induced cardiomyocytes (P < 0.001, )). Opposite to the effects of over-expressed miR-181a-5p, MTT analysis verified that SIRT1 directly increased the viability of H/R-induced cardiomyocytes, and rescued the inhibitory effect of miR-181a-5p on cell viability (P < 0.001, )). After adding SIRT1, the levels of LDH release and MDA of H/R-induced cardiomyocytes were evidently reduced, whereas the level of SOD was greatly increased (P < 0.001, ,f)). As for flow cytometry, although over-expressed miR-181a-5p facilitated the apoptosis of H/R-induced cardiomyocytes, its effect as such could be reversed by SIRT1 (P < 0.001, ,h)). In WB experiments, we found that SIRT1 increased the expressions of Bcl-2 and decreased Bax and Caspase 3 expression in H/R-induced cardiomyocytes, and counteracted the effects of over-expressed miR-181a-5p on apoptosis-related proteins (P < 0.05, ,j)).
Figure 3. SIRT1 counteracted the effects of over-expressed miR-181a-5p on hypoxia/reoxygenation (H/R)-induced cardiomyocytes.
Discussion
Cell apoptosis, which is a process of cell death triggered by physiological or pathological factors, can be regulated by genes [Citation23]. Evidence indicated that myocardial injury caused by I/R showed necrosis and apoptosis of both cardiomyocyte; however, there is no method available to intervene cell necrosis [Citation24]. Cardiomyocytes apoptosis, which is regulated by a series of biological processes, can be reversed through appropriate intervention means [Citation25]. It has been confirmed that miRNAs such as miR-203 can regulate cardiomyocytes apoptosis and has the ability to reduce myocardial tissue injury and cardiomyocytes death caused by myocardial infarction [Citation26]. Moreover, miR-496 can target hook microtubule tethering protein 3 (Hook3) to rescue cardiomyocytes apoptosis caused by H/R through activating PI3K/Akt/mTOR signal transduction pathway [Citation27]. A recent study reported that knocking down miR-181a plays a protective role in myocardium under hypoxia through correlative signaling, thereby alleviating cardiomyocytes apoptosis [Citation28]. Moreover, Wang L et al. [Citation29] also demonstrated that the down-regulation of miR-181a notably inhibits the apoptosis of H9C2 cells induced by hydrogen peroxide through controlling the mitochondrial apoptosis pathway. These previous findings revealed that miR-181a has a critical function in cardiac apoptosis under different induction conditions; however, its effects on H/R-induced cardiomyocyte apoptosis and the mechanisms still remain indistinct.
In the present study, miR-181a-5p was confirmed to have the target site of SIRT1 3ʹ UTR; thus, we speculated that miR-181a-5p regulated SIRT1 to regulate H/R-induced cardiomyocyte proliferation. In later experiments, we first constructed H/R injury cardiomyocyte models and up-regulated or down-regulated miR-181a-5p expressions in the cells. The laboratorial outcomes illuminated that over-expressed miR-181a-5p under H/R injury condition could inhibit the survival ability of cardiomyocytes and accelerate cell apoptosis, while the miR-181a-5p knockdown had completely opposite effects. Sun Y et al. [Citation30] indicated that up-regulated miR-181a-5p suppressed the proliferation of myeloma cells and promoted apoptosis through related signaling pathways. Similarly, Zhu L et al. [Citation31] proved that up-regulated miR-181a-5p increased apoptosis and suppressed amplification of cervical cancer cells. In animal experiments, scholars found that the down-regulation of miR-181a-5p may be related to the inhibition of apoptosis of rat endothelial cells [Citation32]. Thus, miR-181a-5p could increase the apoptosis of various cells, and it plays different roles in different diseases.
During H/R injury, excessive produce of reactive oxygen species (ROS) can cause denaturation of tissue DNA and protein, induce lipid peroxidation, and disturb cytokine levels, thereby ultimately leading to changes in cellular functions and structures and cardiomyocyte apoptosis [Citation33,Citation34]. As a metabolite of lipid peroxidation, MDA can often reflect the severity of cells injured by ROS [Citation35]. SOD is an antioxidant enzyme and can remove ROS, balance oxidation, and anti-oxidation, thus protecting cells from damage [Citation36]. Moreover, LDH is a glycolytic enzyme and can be released to the outside of cells after cell damage; therefore, the degree of cell injury can be evaluated by detecting the amount of released LDH [Citation37]. Our report also disclosed that down-regulated miR-181a-5p reduced the release of LDH and MDA level of cardiomyocytes, and meanwhile increased the SOD level in the cells, indicating that the low-expression of miR-181a-5p has a protective effect on the injury of cardiomyocytes caused by H/R. Furthermore, we also measured the expressions of apoptosis-related proteins, covering Bcl-2, Bax, and Caspase 3, in cardiomyocytes. Thereinto, Bcl-2 is an anti-apoptotic protein, while Bax and Caspase 3 have pro-apoptotic effects [Citation38]. The experimental analysis demonstrated that, opposite to knockdown of miR-181a-5p, over-expressing miR-181a-5p inhibited the expressions of Bcl-2 and Caspase 3 and elevated Bax expression in H/R-induced cardiomyocytes. Thus, these investigations manifested that miR-181a-5p was involved in apoptosis of H/R-induced cardiomyocytes through regulating oxidative stress response and related protein pathways.
Nevertheless, though up-regulating miR-181a-5p had adverse effects on the survival of cardiomyocytes, it should be noted that SIRT1 could reverse its pro-apoptotic and anti-viability effects on H/R-induced cell damage. Zhou L et al. [Citation39] found that SIRT1 over-expression obviously increased the activities of hypoxia-induced osteoblasts and reduced the proportion of apoptosis by reducing the activation of Caspases 3, Caspases 9 and related pathways. Anything else, Wang S et al. [Citation40] demonstrated that SIRT1 up-regulation suppressed hyperglycemia-activated apoptosis in human endothelial cells via alleviating mitochondrial dysfunction and relieving oxidative stress. Previous study indicated that miR-181a promoted the transcription ability of the apoptosis-promoting element Forkhead box O1 (FoxO1) and accelerated apoptosis of granulosa cells by down-regulating SIRT1, which are consistent with the results of our study [Citation41]. In addition, studies showed that SIRT1 is effective in treating cardiac diseases such as diabetic cardiomyopathy [Citation42], cardiovascular disease [Citation43]. Thus, the above results indicated the beneficial effects of SIRT1 on cardiomyocytes damage induced by H/R, and such effects of SIRT1 might be regulated by miR-181a-5p. However, our research is still not deep enough, and further animal models need to be established to verify the role of miR-181a-5p in H/R-induced cardiomyocyte apoptosis by regulating SIRT1 expression.
Conclusions
In conclusion, miR-181a-5p is involved in the H/R-induced cardiomyocytes apoptosis through regulating the expression of SIRT1. Down-regulation of miR-181a-5p can increase H/R-induced cardiomyocyte viability and suppress apoptosis, thus, miR-181a-5p could be explored as a novel therapeutic direction for treating myocardial I/R-related diseases.
Authors’ contributions
Substantial contributions to conception and design: MQ, LH
Data acquisition, data analysis, and interpretation: XM, LH
Drafting the article or critically revising it for important intellectual content: MQ, LH, ZL
Final approval of the version to be published: All authors
Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of the work are appropriately investigated and resolved: ZL.
Ethics approval and consent to participate
No human and animals are involved in this research.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Reed GW, Rossi JE, Cannon CP. Acute myocardial infarction. Lancet. 2017 Jan 14;389(10065):197–210. PubMed PMID: 27502078; eng. .
- Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013 Jan;123(1):92–100. PubMed PMID: 23281415; PubMed Central PMCID: PMCPMC3533275. eng. .
- Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol. 2010 Aug 1;106(3):360–368. PubMed PMID: 20643246; PubMed Central PMCID: PMCPMC2957093. eng. .
- Kalogeris T, Baines CP, Krenz M, et al. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. PubMed PMID: 22878108; PubMed Central PMCID: PMCPMC3904795. eng. .
- Gottlieb RA, Engler RL. Apoptosis in myocardial ischemia-reperfusion. Ann N Y Acad Sci. 2010;874(1):412–426.
- Simon F, Oberhuber A, Floros N, et al. Acute limb ischemia-much more than just a lack of oxygen. Int J Mol Sci. 2018 Jan 26;19(2):374. PubMed PMID: 29373539; PubMed Central PMCID: PMCPMC5855596. eng.
- Ding HS, Yang J, Chen P, et al. The HMGB1-TLR4 axis contributes to myocardial ischemia/reperfusion injury via regulation of cardiomyocyte apoptosis. Gene. 2013 Sep 15;527(1):389–393. PubMed PMID: 23727604; eng.
- Rodrigo R, Fernandez-Gajardo R, Gutierrez R, et al. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 2013 Aug;12(5):698–714. PubMed PMID: 23469845; eng.
- Gottlieb RA. Cell death pathways in acute ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther. 2011 Sep-Dec;16(3–4):233–238. PubMed PMID: 21821521; PubMed Central PMCID: PMCPMC3337030. eng. .
- Li Y, Zhu X, Liu X, et al. miR-200a mediates protection of thymosin beta-4 in cardiac microvascular endothelial cells as a novel mechanism under hypoxia-reoxygenation injury. J Cell Biochem. 2019 Jul 2;120(7):12069. PubMed PMID: 31265170; eng.
- Gao C, Wang R, Li B, et al. TXNIP/Redd1 signaling and excessive autophagy: a novel mechanism of myocardial ischemia/reperfusion injury in mice. Cardiovasc Res. 2019 Jun 26;116(1):e1–e4. PubMed PMID: 31241142; eng.
- Manning JR, Thapa D, Zhang M, et al. Loss of GCN5L1 in cardiac cells disrupts glucose metabolism and promotes cell death via reduced Akt/mTORC2 signaling. Biochem J. 2019 Jun 19;476(12):1713–1724. PubMed PMID: 31138772; eng.
- Jennings RB, Reimer KA. The cell biology of acute myocardial ischemia. Annu Rev Med. 1991;42:225–246. PubMed PMID: 2035969; eng.
- McCall MN, Kim MS, Adil M, et al. Toward the human cellular microRNAome. Genome Res. 2017 Oct;27(10):1769–1781. PubMed PMID: 28877962; PubMed Central PMCID: PMCPMC5630040. eng.
- Mohr AM, Mott JL. Overview of microRNA biology. Semin Liver Dis. 2015 Feb;35(1):3–11. PubMed PMID: 25632930; PubMed Central PMCID: PMCPMC4797991. eng.
- Zhao Y, Ponnusamy M, Dong Y, et al. Effects of miRNAs on myocardial apoptosis by modulating mitochondria related proteins. Clin Exp Pharmacol Physiol. 2017 Apr;44(4):431–440. PubMed PMID: 28008641; eng.
- Qin D, Wang X, Li Y, et al. MicroRNA-223-5p and −3p cooperatively suppress necroptosis in ischemic/reperfused hearts. J Biol Chem. 2016 Sep 16;291(38):20247–20259. PubMed PMID: 27502281; PubMed Central PMCID: PMCPMC5025706. eng.
- Diao H, Liu B, Shi Y, et al. MicroRNA-210 alleviates oxidative stress-associated cardiomyocyte apoptosis by regulating BNIP3. Biosci Biotechnol Biochem. 2017 Sep;81(9):1712–1720. PubMed PMID: 28661226; eng.
- Feng X, Zhang C, Yang Y, et al. Role of miR-181a in the process of apoptosis of multiple malignant tumors: a literature review. Adv Clin Exp Med. 2018 Feb;27(2):263–270. PubMed PMID: 29521071; eng.
- Du M, Zhang Z, Gao T. Piceatannol induced apoptosis through up-regulation of microRNA-181a in melanoma cells. Biol Res. 2017 Oct 17;50(1):36. PubMed PMID: 29041990; PubMed Central PMCID: PMCPMC5644130. eng. .
- Fang YC, Yeh CH. Inhibition of miR-302 suppresses hypoxia-reoxygenation-induced H9c2 cardiomyocyte death by regulating Mcl-1 expression. Oxid Med Cell Longev. 2017;2017(2017):7968905. PubMed PMID: 28491238; PubMed Central PMCID: PMCPMC5405583. eng.
- Rao X, Lai D, Huang X. A new method for quantitative real-time polymerase chain reaction data analysis. J Comput Biol. 2013 Sep;20(9):703–711. PubMed PMID: 23841653; PubMed Central PMCID: PMCPMC3762066. eng.
- Hassan M, Watari H, AbuAlmaaty A, et al. Apoptosis and molecular targeting therapy in cancer. Biomed Res Int. 2014;2014:150845. PubMed PMID: 25013758; PubMed Central PMCID: PMCPMC4075070. eng.
- Miura T, Tanno M, Sato T. Mitochondrial kinase signalling pathways in myocardial protection from ischaemia/reperfusion-induced necrosis. Cardiovasc Res. 2010 Oct 1;88(1):7–15. PubMed PMID: 20562423; eng.
- Li X, Ming Y, Niu Y, et al. [Programmed necrosis: a new target for ischemia reperfusion injury]. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2016 Jul;41(7):765–770. PubMed PMID: 27592584; chi.
- Zhang J, Pan J, Yang M, et al. Up-regulating microRNA-203 alleviates myocardial remodeling and cell apoptosis through down-regulating PTP1B in rats with myocardial infarction. J Cardiovasc Pharmacol. 2019 Aug 10;75(5):474–481. PubMed PMID: 31425379; eng.
- Jin Y, Ni S. miR-496 remedies hypoxia reoxygenation-induced H9c2 cardiomyocyte apoptosis via Hook3-targeted PI3k/Akt/mTOR signaling pathway activation. J Cell Biochem. 2019 Aug 22;121(1):698–712. PubMed PMID: 31436348; eng.
- Zhu J, Yao K, Guo J, et al. miR-181a and miR-150 regulate dendritic cell immune inflammatory responses and cardiomyocyte apoptosis via targeting JAK1-STAT1/c-Fos pathway. J Cell Mol Med. 2017 Nov;21(11):2884–2895. PubMed PMID: 28597963; PubMed Central PMCID: PMCPMC5661264. eng.
- Wang L, Huang H, Fan Y, et al. Effects of downregulation of microRNA-181a on H2O2-induced H9c2 cell apoptosis via the mitochondrial apoptotic pathway. Oxid Med Cell Longev. 2014;2014:960362. PubMed PMID: 24683439; PubMed Central PMCID: PMCPMC3942394. eng.
- Sun Y, Jiang T, Jia Y, et al. LncRNA MALAT1/miR-181a-5p affects the proliferation and adhesion of myeloma cells via regulation of Hippo-YAP signaling pathway. Cell Cycle (Georgetown, Tex). 2019 Aug 9;18(19):2509–2523. PubMed PMID: 31397203; eng.
- Zhu L, Zhang Q, Li S, et al. Interference of the long noncoding RNA CDKN2B-AS1 upregulates miR-181a-5p/TGFbetaI axis to restrain the metastasis and promote apoptosis and senescence of cervical cancer cells. Cancer Med. 2019 Apr;8(4):1721–1730. PubMed PMID: 30884187; PubMed Central PMCID: PMCPMC6488111. eng.
- Zhang Q, Xiao X, Zheng J, et al. A glucagon-like peptide-1 analog, liraglutide, ameliorates endothelial dysfunction through miRNAs to inhibit apoptosis in rats. PeerJ. 2019;7(5):e6567. PubMed PMID: 30863684; PubMed Central PMCID: PMCPMC6408912. eng.
- Li Q, Xiang Y, Chen Y, et al. Ginsenoside Rg1 protects cardiomyocytes against hypoxia/reoxygenation injury via activation of Nrf2/HO-1 signaling and inhibition of JNK. Cell Physiol Biochem. 2017;44(1):21–37. PubMed PMID: 29130959; eng.
- Coimbra-Costa D, Alva N, Duran M, et al. Oxidative stress and apoptosis after acute respiratory hypoxia and reoxygenation in rat brain. Redox Biol. 2017 Aug;12(8):216–225. PubMed PMID: 28259102; PubMed Central PMCID: PMCPMC5334548. eng.
- Tsikas D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: analytical and biological challenges. Anal Biochem. 2017 May 1;524(1):13–30. PubMed PMID: 27789233; eng.
- Bresciani G, da Cruz IB, Gonzalez-Gallego J. Manganese superoxide dismutase and oxidative stress modulation. Adv Clin Chem. 2015;68:87–130. PubMed PMID: 25858870; eng.
- Undyala V, Terlecky SR, Heide RSV. Targeted intracellular catalase delivery protects neonatal rat myocytes from hypoxia-reoxygenation and ischemia-reperfusion injury. Cardiovasc Pathol. 2011;20(5):272–280.
- Tian Y, Du YY, Shang H, et al. Calenduloside E analogues protecting H9c2 cardiomyocytes against H2O2-induced apoptosis: design, synthesis and biological evaluation. Front Pharmacol. 2017;8(23):862. PubMed PMID: 29218010; PubMed Central PMCID: PMCPMC5703861. eng.
- Zhou L, Wang SI, Moon YJ, et al. Overexpression of SIRT1 prevents hypoxia-induced apoptosis in osteoblast cells. Mol Med Rep. 2017 Sep;16(3):2969–2975. PubMed PMID: 28677728; eng.
- Wang S, Wang J, Zhao A, et al. SIRT1 activation inhibits hyperglycemia-induced apoptosis by reducing oxidative stress and mitochondrial dysfunction in human endothelial cells. Mol Med Rep. 2017 Sep;16(3):3331–3338. PubMed PMID: 28765962; eng.
- Zhang M, Zhang Q, Hu Y, et al. miR-181a increases FoxO1 acetylation and promotes granulosa cell apoptosis via SIRT1 downregulation. Cell Death Dis. 2017 Oct 5;8(10):e3088. PubMed PMID: 28981116; PubMed Central PMCID: PMCPMC5680589. eng.
- Karbasforooshan H, Karimi G. The role of SIRT1 in diabetic cardiomyopathy. Biomed Pharmacother. 2017 Jun;90(8):386–392. PubMed PMID: 28380414; eng.
- D’Onofrio N, Servillo L, Balestrieri ML. SIRT1 and SIRT6 signaling pathways in cardiovascular disease protection. Antioxid Redox Signal. 2018 Mar 10;28(8):711–732. PubMed PMID: 28661724; PubMed Central PMCID: PMCPMC5824538. eng.