
ABSTRACT
Chemical screening of culture medium from the soil fungus Stachybotrys sp. resulted in the isolation of the three new phenylspirodrimanes MBJ-0030 (1), MBJ-0031 (2) and MBJ-0032 (3). Their structures were determined by detailed analysis of spectroscopic data. The absolute configurations of 1–3 were determined by modified Mosher’s and Marfey’s methods. In addition, cytotoxic and antimicrobial evaluations of the compounds were conducted.
GRAPHICAL ABSTRACT

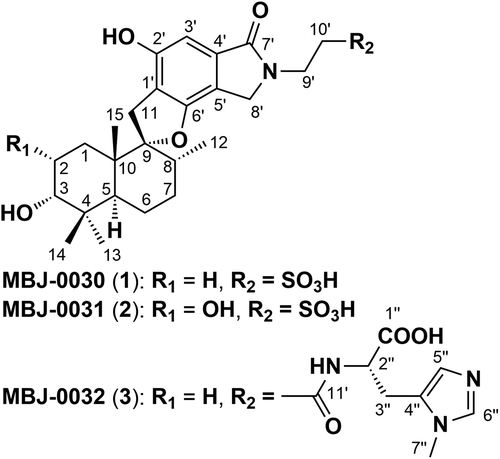
The structure of new phenylspirodrimane metabolites MBJ-0030, MBJ-0031, and MBJ-0032 isolated from the soil fungal strain Stachybotrys sp. f23793.
Over the past decade we have constructed a library of isolated natural products and with which we have executed a range of biological screening efficiently [Citation1]. The novel compounds in our library, series name “JBIR”, were obtained by chemical or biological screening from our in-house microorganism strain collections. We recently began to expand and develop this library by investigating rare microbial products using an advanced compound identification system based on accumulated HPLC-MS profiling data combined with strain information. This new library was designated “MBJ’s special selection” and has provided a variety of compounds (the “MBJ series”) from various microbial culture [Citation2–Citation7]. Further chemical screening for structurally intriguing compounds led to the identification of MBJ-0030 (1), MBJ-0031 (2), and MBJ-0032 (3) from the fermentation broth of Stachybotrys sp. (). Herein, we describe the fermentation, isolation, structural elucidation, and preliminary biological activities of 1–3.
Figure 1. Structures of 1–3.
Material and methods
General
Optical rotations were measured on a HORIBA SEPA-300 high sensitive polarimeter using a 589 nm wavelength sodium lamp. UV and IR spectra were measured with a Beckman Coulter DU730 UV/Vis spectrophotometer and a HORIBA FT-720 spectrometer, respectively. Both 1D and 2D NMR spectra were measured using a Varian NMR System 600 NB CL or 500 NB CL with MeOH-d4 (δC 49.0, δH 3.31 ppm) as the solvent and the residual solvent peak served as the internal standard. The coupling constants (J) are given in hertz. HR-ESIMS data were recorded using a Waters LCT-Premier XE mass spectrometer. Normal- and reversed-phase MPLC separations were carried out using Purif-Pack Si-30 and Purif-Pack ODS-100 columns (Shoko Scientific Co., Ltd.), respectively. Gel filtration was performed using a Sephadex LH-20 column (GE Healthcare Bio-Sciences AB). Analytical RP-UPLC was performed using a Waters ACQUITY UPLC System in conjunction with a BEH ODS column (2.1 i.d. × 100 mm, Waters), a Waters ACQUITY UPLC photodiode array eλ detector, and an LCT-Premier XE mass spectrometer.
Strain isolation and identification
The fungal strain Stachybotrys sp. f23793 was isolated from a soil sample collected in early October 2005 in Shizuoka prefecture, Japan. The identification of the fungal strain was accomplished on the basis of its D1/D2 domain in the 28S rDNA sequence, which gave 98.5% sequence similarity to S. echinata using the BLASTN search tool.
Extraction and isolation
The strain was cultivated in 250-mL Erlenmeyer flasks, each containing 25 mL of seed medium consisting of 2% potato starch, 1% glucose, 2% soybean powder, 0.1% KH2PO4, and 0.05% MgSO4·7H2O. The flasks were shaken on a rotary shaker (220 rpm) at 25°C for 3 days. Aliquots (0.5 mL) of the broth were transferred to 500-mL Erlenmeyer flasks containing 50 mL of production medium consisting of 2% potato starch, 1% glucose, 2% soybean powder, 0.1% KH2PO4, and 0.05% MgSO4·7H2O and were cultured on a rotary shaker (220 rpm) at 25°C for 4 days.
The entire culture broth (2 L) was extracted with an equal volume of n-BuOH. The organic layer was evaporated and the resultant extract was then diluted with brine (300 mL), washed with EtOAc (300 mL × 3), and extracted with n-BuOH (300 mL × 2) successively. The n-BuOH extract (3.63 g) was applied to an ODS-MPLC column (Purif-Pack ODS-100) and developed with 40–100% MeOH in H2O by a stepwise 20% gradient. The 60% MeOH eluate (642 mg) was re-chromatographed by ODS-MPLC (Purif-Pack ODS-100; 40–100% aq. MeOH, 20% stepwise) and the obtained 60% MeOH eluate (287 mg) was chromatographed by ODS-MPLC (Purif-Pack ODS-30; 10–100% aq. CH3CN, 10% stepwise, UV detection at 254 nm) to afford 1 (11.6 mg) and crude 2 (246.3 mg). The purification of the 2 was carried out by ODS-MPLC (Purif-Pack ODS-30; 20, 25, 30, and 100% CH3CN in H2O, stepwise, UV detection at 254 nm) to yield 49.4 mg 2. The 80% MeOH fraction (546 mg) of the n-BuOH extract was subjected to ODS-MPLC (Purif-Pack ODS-30; 40–100% aq. MeOH, 10% stepwise) to obtain crude 3 (115.5 mg). Further purification was conducted by ODS-MPLC (Purif-Pack ODS-30; 20–100% aq. CH3CN, stepwise, UV detection at 254 nm) followed by gel filtration on a Sephadex LH-20 column (CHCl3/MeOH = 1:1) to yield 3 (29.5 mg).
MBJ-0030 (1)
Colorless amorphous powder; [α]24D – 38 (c 0.05, MeOH); UV (MeOH) λmax (logε) 264 (3.9), 299 (3.4), and 302 (3.4) nm; IR (ATR) νmax: 3400 (hydroxy) and 1631 (carbonyl) cm−1; 1H NMR (600 MHz) and 13C NMR (150 MHz), see ; HRESIMS m/z at 492.2079 [M – H] – (calcd for C25H34NO7S, 492.2056).
Table 1. 13C and 1H NMR spectroscopic data for MBJ-0030 (1), MBJ-0031 (2), and MBJ-0032 (3) in CD3OD.
MBJ-0031 (2)
Colorless amorphous powder; [α]24D – 65 (c 0.05, MeOH); UV (MeOH) λmax (logε) 264 (3.9), 299 (3.5), and 302 (3.5) nm; IR (νmax (ATR) 3400 and 1625 cm–1); 1H NMR (500 MHz) and 13C NMR (125 MHz), see ; HRESIMS m/z at 508.2028 [M–H] – (calcd for C25H34NO8S 508.2005).
MBJ-0032 (3)
Colorless amorphous powder; [α]24D – 30 (c 0.05, MeOH); UV (MeOH) λmax (logε) 264 (3.9), 299 (3.4), and 302 (3.5) nm; IR (νmax (ATR) 3400 and 1625 cm–1); 1H NMR (600 MHz) and 13C NMR (150 MHz), see ; HRESIMS m/z at 607.3144 [M–H]–, (calcd for C33H43N4O7 607.3132).
Methylation of 1
To a MeOH solution (300 μL) of 1 (2.2 mg) was treated with trimethylsilyldiazomethane (10% in hexane, 200 μL), and the mixture was stirred at room temperature overnight. Removal of the solvent under a gentle air stream gave a methyl ester derivative of 1 (2.6 mg).
Esterification of methyl ester derivative of 1 with MTPA chloride
To a pyridine-d5 solution (450 μL) of methyl ester derivative of 1 (1.3 mg) was added (R)- or (S)-MTPA chloride (12 μL) at room temperature for 2 days, and the 1H NMR spectrum was recorded for the (S)- or (R)-MTPA ester.
Marfey’s analysis
Compound 3 (0.4 mg) was hydrolyzed in 6N HCl (0.45 mL) at 110°C for 16 h. After acid hydrolysis, the reaction solution was evaporated in vacuo. The residue was dissolved in 0.1 M NaHCO3 (0.1 mL) and 1% N-(5-fluoro-2,4-dinitrophenyl)-l-alaninamide (FDAA) in acetone (0.1 mL) was added. The mixture was then heated at 40°C for 30 min. The reaction products were analyzed using a HPLC-MS system; after sample injection onto a Capcell Pak C18 MGII column (4.6 i.d. × 150 mm), the column was developed using a linear gradient of aqueous CH3CN containing 0.1% formic acid (10–30% CH3CN in 15 min, flow rate 1.0 mL/min). The FDAA derivatives were detected by ion-selective monitoring at m/z 422 [M+H]+.
Results and discussion
Culture broth of Stachybotrys sp. was extracted with n-BuOH and concentrated in vacuo. The residual pellet was partitioned between EtOAc/H2O, followed by n-BuOH extraction. The n-BuOH layer was concentrated and subjected to a combination of chromatographic separations to afford compounds 1–3.
The structures of 1–3 were determined by analysis of HRESIMS, UV absorption, and a series of 2D NMR data sets, including DQF-COSY, HSQC, and CT-HMBC [Citation8]. 13C and 1H NMR data for 1–3 are listed in .
MBJ-0030 (1) was isolated as a colorless amorphous powder that yielded an [M – H] – ion at m/z 492.2079 in HRESIMS, which corresponded to the molecular formula C25H35NO7S (calcd for C25H34NO7S, 492.2056). Analysis of the DQF-COSY spectrum indicated the presence of four 1H sequences from a doublet methyl proton 12-H3 (δH 0.73) through a methine proton 8-H (δH 1.85) to methylene protons 7-H2 (δH 1.59, 1.54), between a methine proton 5-H (δH 2.13) and methylene protons 6-H2 (δH 1.58, 1.51), between methylene protons 1-H2 (δH 1.82, 1.06) and methylene protons 2-H2 (δH 1.95, 1.52), and between nitrogen-substituted methylene protons 9′-H2 (δH 4.00) and methylene protons 10′-H2 (δH 3.16). The 1H chemical shift values of 6-H and 7-H were too crossed to determine the connection between these protons. The CT-HMBC spectrum showed 1H–13C long-range correlations from singlet geminal-methyl protons 13-H3 (δH 0.98) and 14-H3 (δH 0.89) to an oxymethine carbon C-3 (δC 76.4), a quaternary carbon C-4 (δC 38.7), and a methine carbon C-5 (δC 41.3), and from a singlet methyl proton 15-H3 (δH 1.05) to C-5, a quaternary carbon C-10 (δC 43.5) and an aliphatic methylene carbon C-1 (δC 25.4) which in turn was coupled to an oxymethine proton 3-H (δH 3.33). These correlations proved the presence of a 2,2,4-trimethylcyclohexanol moiety ()).
Figure 2. (a) NMR analysis of 1. COSY: bold line; HMBC (1H to 13C): arrows. (b) Relative configurations of drimane sesquiterpenoid moiety of 1 (NOESY: arrow). (c) ΔδS–R values for MTPA derivatives of the methyl ester derivative of 1. (d) NMR analysis of 2. COSY: bold line; HMBC (1H to 13C): arrows. (e) Relative configurations of drimane sesquiterpenoid moiety of 2 (NOESY: arrow). (f) NMR analysis of 3. COSY: bold line; HMBC (1H to 13C): arrows.
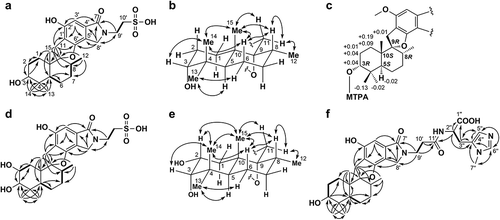
Further HMBC correlations from 12-H3 and 15-H3 to an oxygenated quaternary carbon C-9 (δC 99.7) and from the methylene protons 11-H2 (δH 3.21, 2.84) to a methine carbon C-8 (δC 38.4), C-9 and C-10 appeared to show a consecutive carbon sequence from C-8 to C-10 through C-9, which was directly connected to C-11. The structure of a drimane sesquiterpenoid moiety (C-1 to C-15) was established by HMBC correlations from 5-H to the aliphatic carbons C-6 (δC 22.1) and C-7 (δC 32.2). The remaining substructure was established as follows. Strong 1H–13C long-range couplings were observed from an aromatic proton 3′-H (δH 6.66) to quaternary aromatic carbons C-1′ (δC 118.8) and C-5′ (δC 114.6) at meta-positions, an amide carbonyl carbon C-7′ (δC 171.0) at the peri-position, and a weak coupling to an oxygenated aromatic quaternary carbon C-2′ (δC 155.1). Additionally, the nitrogen-substituted methylene protons 8′-H2 (δH 4.55, 4.47; δC 49.0) were correlated with C-5′, an aromatic quaternary carbon C-4′ (δC 134.9), an oxygenated aromatic quaternary carbon C-6′ (δC 157.5), and C-7′. These results indicated the presence of a 4,6-dihydroxy-1-isoindolinone moiety. Direct connectivity between C-11 and C-1′ was revealed by 1H–13C long-range correlations from 11-H2 to C-1′, C-2′, and C-6′. Additional HMBC correlations from 9′-H2 to C-7′ and C-8′, together with consideration of the 1H and 13C NMR chemical shifts of the methylene unit C-10′ (δH 3.16, δC 50.5) [Citation9] and the molecular formula of 1, suggested that an ethanesulfonic acid group was substituted at a nitrogen atom of the 4,6-dihydroxy-1-isoindolinone moiety. The degree of unsaturation of 1 requires an additional ring structure and we confirmed the presence of a phenolic hydroxy group by methylating 1 with trimethylsilyldiazomethane in MeOH. The substituted position of the methyl group was confirmed by an HMBC correlation from the emerging methoxy proton (δH 3.86 in MeOH-d4) to the oxygenated aromatic carbon C-2′ (δC 157.8) and a NOESY correlation between the methoxy proton and the aromatic methine proton 3′-H (δH 6.82). These facts suggested the presence of a hydroxy group at the C-2′ position in 1, whereas an oxygen atom at C-6′ is aprotonic (constructing ether bond). Therefore, the gross structure of 1 was concluded to be as shown in .
The relative configuration of 1 was established by NOESY spectra ()). A NOESY correlation between an axial proton 5-H (J = 2.4, 12.0 Hz) and 13-H3 implied that 13-CH3 is in an equatorial orientation. The axial orientations of 3-OH and 15-CH3 were revealed from NOESY correlations between 14-H3 and 3-H and 15-H3. Furthermore, 15-H3 was also correlated to 8-H and 11-Ha (δH 3.21), which indicated that C-11 and C-12 are in equatorial orientations. Thus, the relative configuration of 1 was established to be as shown in .
The absolute configuration of C-3 was assigned using the modified Mosher method [Citation10,Citation11]. After O-methylation of the phenolic hydroxy group at C-2′ with trimethylsilyldiazomethane in MeOH, treatment of the methyl ester derivative of 1 with (R)- and (S)- methoxy(trifluoromethyl)phenylacetic acid (MTPA) chloride in dry pyridine afforded the (S)- and (R)-MTPA esters, respectively. The difference in chemical shift values (Δδ = δS – δR) was calculated to assign the 3R configuration to 1 ()). By taking into consideration their relative configurations, C-5, C-8, C-9 and C-10 were interpreted to be in the S, R, R and S configurations, respectively.
The UV absorption maxima (logε) in MeOH at 264 (3.9), 299 (3.4), and 302 (3.5) nm and the IR spectrum (νmax: 3400 and 1625 cm−1) of 2 implied that 2 is a congener of 1. The molecular formula of 2 was established as C25H35NO8S by HRESIMS data (obsd m/z 508.2028 [M – H]–, calcd for C25H34NO8S 508.2005). The 1H and 13C NMR chemical shifts of 2 were similar to those of 1 (), except for the C-2 moiety. The chemical shifts of C-2 (δH 3.94, δC 67.2) and the molecular formula of 2 suggested the presence of an additional hydroxy group at the C-2 position. Detailed analyses of the 2D NMR data supported the proposed structure shown in ). The partial relative configuration of 2 was determined by NOESY to be the same as 1 ()). Additionally, the large coupling constant value (12.0 Hz) between 2-H and 1-Ha (δH 1.75) indicated that 2-H and 1-Ha are in diaxial orientations and that the hydroxy group at C-2 is in the equatorial orientation. NOESY correlations among 14-H3, 15-H3, and 2-H supported this orientation. From the absolute configuration of 1, the absolute configuration of 2 was established as 2R, 3S, 5S, 8R, 9R and 10S.
MBJ-0032 (3) features the following properties: [α]24D – 30 (c 0.05, MeOH); UV λmax (logε) in MeOH: 264 (3.9), 299 (3.4), and 302 (3.5) nm; HRESIMS: m/z 607.3144 [M–H]–, calcd for C33H43N4O7 607.3132; and IR absorption (νmax): 3400 and 1625 cm–1. The NMR spectra indicated that a partial structure of 3 was the same as that of 1, including the relative configuration, with the exception of an anserine moiety as described below. 1H spin couplings between an α-methine proton 2′′-H (δH 4.52) and methylene protons 3′′-H2 (δH 3.19, 2.98) and 1H–13C long-range couplings from 2′′-H to a carbonyl carbon C-1′′ (δC 175.8) and an aromatic quaternary carbon C-4′′ (δC 132.8), from 3′′-H2 to C-1′′, C-4′′, and an aromatic methine carbon C-5′′ (δC 121.1) were observed. Furthermore, HMBC correlations from 5′′-H (δH 7.08) to an aromatic methine carbon C-6′′ (δC 137.0), and from the N-methyl proton 7′′-H3 (δH 3.75) to C-4′′ and C-6′′ were observed. Thus, the planar structure of a 3-methylhistidine (synonyms: Nπ-methylhistidine, 3-N-methylhistidine) moiety was revealed to be as shown in ). The structure of a β-alanine moiety was deduced from 1H coupling between nitrogen-bonded methylene protons 9′-H2 (δH 3.77) and the methylene protons 10′-H2 (δH 2.58), together with 1H–13C long-range correlations from 9′-H2 and 10′-H2 to an amide carbonyl carbon C-11′ (δC 172.9), which in turn showed a long-range correlation to 2′′-H. Taken together, these couplings implied the presence of an anserine moiety. Additionally, HMBC correlations from 9′-H2 to a carbonyl carbon C-7′ (δC 171.0) and a nitrogen-substituted methylene carbon C-8′ (δC 49.0) proved that the connectivity from C-9′ to C-7′ and C-8′ was mediated by a nitrogen atom. Hence, the planar structure of 3 was established to be as shown in .
The absolute configuration of the 3-methylhistidine moiety was established using Marfey’s method [Citation12]. N-(5-fluoro-2,4-dinitrophenyl)-l-alaninamide (FDAA) derivatives were identified by comparing their retention times, molecular formulae and UV spectra with those of standard amino acids derived from FDAA-conjugated compounds. The retention times of authentic FDAA histidine derivatives were 9.9 min (3-methyl-d-His) and 10.5 min (3-methyl-l-His). FDAA-conjugated 3-methyl-l-His in the hydrolyzate of 3 showed essentially the same retention time as that of the 3-methyl-l-His authentic sample (10.4 min). Therefore, the structure of 3 was determined to be as shown in ).
The structures of 1–3 were similar to those of chartarlactams which exhibited lipid-lowering effects in HepG2 cells as assessed by Oil Red O staining, and also resemble those of F-1839s, the inhibitor of pancreatic cholesterol esterase isolated from Stachybotrys sp. [Citation13–Citation19]. However, 1–3 did not inhibit of lipid droplet accumulation using AO cell and 3T3-L1 cell up to 50 μM. These compounds were tested for their cytotoxicity against the human ovarian adenocarcinoma cell line SKOV-3 and for their antibacterial activity against Micrococcus luteus. These assays showed that 1–3 are not cytotoxic to SKOV-3 cells (IC50 > 100 μM) and did not exert antibacterial activity against M. luteus (IC50 > 100 μM). The structures of 1–3 are quite unique and will be applied for a variety of screenings in the future.
To conclude, this study led the isolation of three new phenylspirodrimanes. To our knowledge, 1 and 2 are the first examples of phenylspirodrimanes containing a sulfoethyl moiety, and 3 is the first reported phenylspirodrimane containing a 3-methylhistidine unit.
Author contributions
T. Kawahara and M. Itoh conceived and designed the experiments. T. Kawahara and N. Kagaya performed the essential experiments and analyzed the data. T. Kawahara and K. Shin-ya wrote the paper. All authors discussed the results and commented on the manuscript.
Supporting_Information_MBJ-0030_BBB.pdf
Download PDF (3 MB)Acknowledgments
This work was supported in part by the grant “Project focused on developing key technology of discovering and manufacturing drug for next-generation treatment and diagnosis” from the Ministry of Economic, Trade and Industry (METI), and a Grant-in-Aid for Scientific Research (26221204) from the Japan Society for the Promotion of Science (JSPS).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Kawahara T, Nagai A, Takagi M, et al. JBIR-137 and JBIR-138, new secondary metabolites from Aspergillus sp. fA75. J Antibiot. 2012;65:535–538.
- Kawahara T, Itoh M, Izumikawa M, et al. New hydroxamate metabolite, MBJ-0003, from Micromonospora sp. 29867. J Antibiot. 2013;67(3):261–263.
- Kawahara T, Itoh M, Izumikawa M, et al. Cytotoxic sesquiterpenoids MBJ-0009 and MBJ-0010 from a saprobic fungus Nectria sp. f26111. J Antibiot. 2013;66(9):567–569.
- Kawahara T, Itoh M, Izumikawa M, et al. Three eremophilane derivatives, MBJ-0011, MBJ-0012 and MBJ-0013, from an endophytic fungus Apiognomonia sp. f24023. J Antibiot. 2013;66(5):299–302.
- Kawahara T, Itoh M, Izumikawa M, et al. New chaetoglobosin derivatives, MBJ-0038, MBJ-0039 and MBJ-0040, isolated from the fungus Chaetomium sp. f24230. J Antibiot. 2013;66(12):727–730.
- Kawahara T, Itoh M, Izumikawa M, et al. MBJ-0086 and MBJ-0087, new bicyclic depsipeptides, from Sphaerisporangium sp. 33226. J Antibiot. 2014;68:67–70.
- Kawahara T, Itoh M, Izumikawa M, et al. Novel aziridine-containing peptides MBJ-0034 and MBJ-0035 from Streptosporangium sp. 32552. J Antibiot. 2014;67(8):577–580.
- Furihata K, Seto H. Constant time HMBC (CT-HMBC), a new HMBC technique useful for improving separation of cross peaks. Tetrahedron Lett. 1998;39(40):7337–7340.
- Horiuchi M, Ohnishi K, Iwase N, et al. A novel isoindoline, porritoxin sulfonic acid, from Alternaria porri and the structure-phytotoxicity correlation of its related compounds. Biosci Biotechnol Biochem. 2003;67:1580–1583.
- Dale JA, Mosher HS. Nuclear magnetic resonance enantiomer reagents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and α-methoxy-α-trifluoromethylphenylacetate (MTPA) esters. J Am Chem Soc. 1973;95:512–519.
- Ohtani I, Kusumi T, Kashman Y, et al. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J Am Chem Soc. 1991;113:4092–4096.
- Marfey P. Determination of d-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res Commun. 1984;49:591–596. .
- Kamalov LS, Aripova SF, Isaev MI. Low-molecular-mass metabolites of fungi I. Stachybotrin from Stachybotrys alternans. Chem Nat Compd. 1997;33:462–468.
- Kamalov LS, Aripova SF, Tashkhodzhaev BT, et al. Low-molecular-weight metabolites of fungi. II. Refinement of the structure of stachybotrin. Chem Nat Compd. 1998;34(5):605–608.
- Kamalov LS, Aripova SF, Isaev MI. Low-molecular-mass metabolites of fungi. III. Stachybotrolide from Stachybotrys alternans. Chem Nat Compd. 1998;34(5):616–619.
- Kamalov LS, Aripova SF, Isaev MI. Low-molecular-mass metabolites of fungi IV. The structures of stachybotrin A and stachybotral. Chem Nat Compd. 1999;35:82–85.
- Sakai K, Watanabe K, Masuda K, et al. Isolation, characterization and biological activities of novel triprenyl phenols as pancreatic cholesterol esterase inhibitors produced by Stachybotrys sp. F-1839. J Antibiot. 1995;48:447–456.
- Zhou D, Li L, Qi H, et al. A new stachybotrin congener from a soil fungus Stachybotrys parvispora strain HS-FG-843. J Antibiot. 2015;68:339–341.
- Li Y, Wu C, Liu D, et al. Chartarlactams A-P, phenylspirodrimanes from the sponge-associated fungus Stachybotrys chartarum with antihyperlipidemic activities. J Nat Prod. 2014;77:138–147.