
ABSTRACT
Previous studies suggest an association of cardiac microvascular endothelial cells (CMECs) hyperpermeability with sepsis-related cardiac injury. Our results showed that CMECs permeability was dependent upon concentration and time of lipopolysaccharides (LPS) stimulation. Integrin ανβ3 expression decreased after LPS stimulation. Pretreatment with anti-integrin ανβ3 antibody enhanced LPS-induced hyperpermeability. Upregulation of integrin ανβ3 decreased LPS-induced hyperpermeability. F-actin remodeling was enhanced after LPS stimulation and was inhibited by up-regulation of integrin ανβ3. Inhibition of Src or Rac1 reduced CMECs permeability after LPS stimulation, but there were no differences in the phosphorylation of Src and Rac1 when over-expressing or blocking integrin β3. After pretreatment with Src or Rac1 inhibitor, no significant difference was found in the expression of integrin ανβ3 in LPS-induced CMECs. These finding suggested that integrin ανβ3 overexpression decreased LPS-stimulated CMECS permeability by inhibition of cytoskeletal remodeling, but the mechanism might not be mediated via Src/Rac1 signaling.
GRAPHICAL ABSTRACT
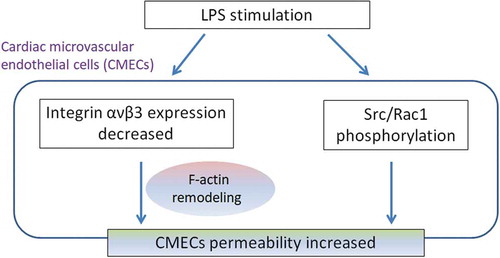
Integrin ανβ3 decreased LPS-stimulated CMECS permeability by inhibition of cytoskeletal remodeling.
Sepsis is a life-threatening condition and is the manifestation of the immune and inflammatory response triggered by infections involving various organs [Citation1]. Sepsis is now the leading cause of death in intensive care units (ICU), accounting for about 10% of all admissions to ICUs [Citation2]. Cardiac dysfunction, the most relevant common complication of severe sepsis, increases the mortality of sepsis. In recent years, the incidence of sepsis-associated cardiac dysfunction has been increasing rapidly. Indeed, approximately 200,000 cases in-hospital cardiac arrests are reported annually in the United States, 15–30% occurring in septic patients [Citation3].
Sepsis causes release of endotoxins by Gram-negative bacteria, triggering an immune response [Citation4]. Lipopolysaccharides (LPS), an endotoxin abundantly released from Gram-negative bacteria, is recognized as the most effective microbial factor in the pathogenesis of sepsis [Citation5]. Endothelial cells as one of the first cell lines against pathogens, can rapidly release inflammatory mediators [Citation6]. In cardiac microvascular endothelial cells (CMECs), exposure to LPS can result in CMECs injury, leading to vascular hyperpermeability [Citation6]. However, the underlying mechanism of sepsis-induced CMEC hyperpermeability is still unclear.
Integrins are transmembrane receptors located on the cell surface as heterodimers of non-covalently associated α and β subunits. There are 18 α subunits and 8 β subunits, resulting in 24 different functional integrins [Citation7]. Integrin αvβ3 is highly expressed in angiogenic endothelial cells and lowly expressed on the surface of resting endothelial cells. The cytoplasmic tail of integrin β3 subunit plays a pivotal role in signal transduction via interaction with proteins. Thus, the β3 subunit is a functional subunit of integrin αvβ3 [Citation8]. The arginine-glycine-aspartic acid peptide abrogates the function of integrin ανβ3, leading to increased permeability of endothelial cell monolayers and isolated porcine coronary venules [Citation9]. β3-knockout mice show increased systemic vascular leakage and mortality [Citation10]. However, the role of integrin αvβ3 in LPS-induced hyperpermeability in CMECs remains unknown.
Therefore, the aim of the present study was to investigate the role of integrin ανβ3 in regulating LPS-induced hyperpermeability of CMECs, and its underlying mechanisms. Our work might provide new targets for the prevention and treatment of sepsis-induced cardiac injury. A better management of cardiac injury during sepsis could improve the survival of patients in the ICU.
Method
Isolation of CMECs
Four-week-old Sprague-Dawley male rats weighing 100 ± 20 g were provided by the Experimental Animal Center, Zhejiang Province. The experimental protocol was performed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1985) and the ARRIVE guideline [Citation11]. The procedures for the care and treatment of animals were approved by the Care of Experimental Animals Committee of Zhejiang Hospital.
Rats were euthanized by cervical dislocation and placed in 70% ethanol for 5 min. The hearts were rapidly excised after thoracotomy and rinsed with ice-cold phosphate buffered saline (PBS) under sterile condition. CMECs were then obtained from the left ventricle, as previously described [Citation12]. Briefly, the left ventricular tissues were finely minced and placed in a 3.5-cm culture dish, which was humidified with 100 μL of fetal bovine serum (FBS; Hyclone, Logan, USA). Tissue pieces were then cultured under humidified air with 5% CO2 at 37°C for 4 h. Once the pieces were firmly attached to the culture dish, 2 mL of M199 (Gibco, Grand Island, USA) supplemented with 20% FBS, 50 mg/L heparin, and 5 ng/mL vascular endothelial growth factor 165 (VEGF165; PeproTech, New Jersey, USA) were added and kept until abundant polygonal oyster-like cells crept out of the myocardial tissues. After removal of tissue pieces and non-adherent cells by washing with ice-cold PBS, 2 mL of fresh culture medium were added for continuous culture. When cells reached 80–90% confluence, cells were digested with 0.1% trypsin (Gino biomedical technology co., LTD, Hangzhou, China) and passaged.
Identification of CMECs
Cells at the second passage were subjected to cytological identification by immunocytochemistry and in vitro tubule formation experiment. Immunocytochemistry was used to detect the expression of factor VIII and CD31. A sterile glass plate was placed in a 24-well plate, and each section was fixed with 4% paraformaldehyde at room temperature for 10 min. After cells reached 70–80% confluence at the bottom of the glass, 0.3% TritonX-100 was used for permeabilization at room temperature for 20 min, followed by inactivation of endogenous peroxidase with 3% H2O2 for 20 min. Each section was blocked with 10% normal goat serum at 37°C for 30 min, and the serum was removed without washing. Each section was incubated with factor VIII polyclonal antibody (diluted with 10% normal goat serum, 1:200 dilution, Abcam, Cambridge, MA, USA) at 4°C for 16 h and CD31 monoclonal antibody (diluted with 10% normal goat serum, 1:200 dilution, Abcam, Cambridge, MA, USA) at 37°C for 3 h. The cells were incubated with horseradish peroxidase-labeled secondary antibody (diluted with PBS, 1:200 dilution, Merck Millipore, Billerica, MA, USA) at 37°C for 30 min, before diaminobenzidine (DAB) development and hematoxylin re-staining. After ethanol gradient dehydration, xylene transparency and neutral gum mounting, the slices were observed under a microscope. The target cells were stained positive for factor VIII and CD31, while the cells in control group had negative staining.
The in vitro tubule formation experiment was conducted using Matrigel (BD Biosciences, San Jose, CA, USA) melted into a liquid overnight at 4°C. A 96-well plate and 200 μL pipette tips were pre-cooled for 30 min. PBS was added to the wells on the edge of the 96-well plate to prevent the edge effect and Matrigel (40 μL per well) was added into the middle wells, followed by gently up and down shaking. The plate was placed on ice for 2 min to make sure even plating before incubation in the incubator for 30 min to solidify Matrigel. The medium was replaced with M199 medium containing 1% FBS, 100 units/mL penicillin, 100 mg/mL streptomycin and HEPES, and the cells were re-suspended and adjusted to 2 × 105/mL. Cells (100 μL per well) were inoculated in the plate, which was placed in the incubator for 16 h. The cells were observed under inverted phase contrast microscope, showing the growth of reticular three-dimensional structures.
LPS stimulation of CMECs
The cells from the second to fourth passages were used in the following experiments. CMECs were seeded at a density of 5 × 105/cm2 on gelatin-coated microporous membranes of a 12-well plate (Corning Costar, NY, USA), and starved in M199 culture medium without FBS for 12 h, followed by addition of a gradient final concentration of 10, 20, 50, 100, 1000, and 2000 ng/mL of LPS (Sigma-Aldrich, St. Louis, MO, USA) for stimulation for 6 h or followed by addition of a final concentration of 50 ng/mL LPS for gradient stimulation of 3, 6, 9, 12, and 18 h. The absence of LPS was considered as a negative control, while the group without cells was considered as a positive control. A triplicate set of wells was used for each group.
Cell treatments
For β3 overexpression, cells were seeded on a 12-well plate at a density of 5 × 105/cm2. When reaching 30–40% confluence, the culture medium was changed and supplemented with integrin β3-overexpressing adenovirus (Shengbo Biological Technology Company, Shanghai, China) at MOI of 100, followed by culturing for 24 h. The null green fluorescent protein virus (Shengbo Biological Technology Company, Shanghai, China) served as control, and integrin β3 overexpression was determined by western blotting (data not shown).
For β3 inhibition, β3 subunit specific antibody (Cell Signaling Technology, Danvers, MA, USA) at a dilution of 1:50 was used. After blocking for 1 h, 50 ng/mL LPS was added and the cells were stimulated for an additional 6 h.
For Rac1 and Src inhibition, the CMECs monolayers were pretreated with 50 μmol/L Rac1 inhibitor N23766 (Tocris Bioscience, Bristol, UK) or with 10 μmol/L Src inhibitor PP2 (Selleck Chemicals, Houston, TX, USA) for 1 h, followed by 50 ng/mL LPS simulation for 6 h. DMSO was used as a negative control.
Endothelial monolayer permeability by FITC-labeled albumin leakage
The assay was performed as previously described [Citation13]. Cells were inoculated at 1 × 105 cells/cm2 on a 1% gelatin-coated transwell chamber membrane. 1.5 mL of culture medium was added to the lower chamber. Cells were cultured in the upper chamber in a 37°C 5% CO2 cell incubator. Cell monolayers are about 95% fused, starved with serum-free culture medium for 12 h, and washed with PBS. Ten milligrams of fluorescein isothiocyanate (FITC)-dextran-albumin (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in and diluted with 5 mL of PBS. Add 1.5 mL PBS and 0.5 mL solution containing FITC-dextran-albumin in the lower chamber, and the cells were incubated in the dark for 45 min. Samples of 100 μL were harvested from the apical and basolateral compartments. The samples were transferred into a black 96-well plate to determine fluorescence intensity using a microplate reader (λex = 490 nm, λem = 520 nm). The permeability (Pa) of FITC-dextran in the endothelial cell monolayer was calculated as follows: Pa=[A]/t × 1/A × v/[L], where [A] denotes the protein concentration in the apical compartment (calculated by fluorescent intensity), t is the time (seconds), A represents the surface area of the microporous membrane (cm2), v denotes the volume of liquid and [L] refers to the protein concentration in the basolateral compartment. Results were presented by the percentage changes in Pa (Pa% = (Pa of test sample/Pa of control) ×100), and each experiment was performed in triplicate.
Western blotting
Total cell protein was extracted with TRIZAL (Gibco-BRL, Gaithersburg, USA) and the protein concentration was measured using the Bradford method. Then, 20 μg of protein were boiled at 98ºC for 5 min. Tris-Glycine SDS-polyacrylamide gel electrophoresis was performed, followed by transfer to polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA, USA). The blots were probed with antibodies against integrin ανβ3 (1:50 dilution; Cell Signaling Technology, Danvers, MA, USA), total Src, phospho(Ser17)-Src (1:50 dilution; Cell Signaling Technology, Danvers,MA, USA), total Rac1, and phospho(Ser71)-Rac1 (1:200 dilution; Merck Millipore, Billerica, MA, USA). Horseradish peroxidase-conjugated secondary antibodies (1:200 dilution; Merck Millipore, Billerica, MA, USA) were used in conjunction with an enhanced chemiluminescence (ECL) detection system (Amersham, UK).
Immunofluorescence
The cells were fixed with 4% paraformaldehyde and permeabilized with 0.25% Triton X-100 for 30 min. After blocking with 10% normal goat serum at room temperature for 1 h, cells were incubated with anti-integrin ανβ3 monoclonal antibody (1:50 dilution; Abcam, Cambridge, MA, USA) for 16 h at 4ºC, followed by Alexa Fluor 568 conjugated antibody (1:200 dilution; Invitrogen, Carlsbad, CA, USA) and 1% rhodamine-phalloidin (Invitrogen, Carlsbad, CA, USA) for 1 h at 37ºC, respectively. The nuclei were counterstained with DAPI at room temperature. The fluorescent images were obtained with a confocal scanning laser microscope (Zeiss, Germany).
Statistical analysis
All data were expressed as mean ± standard error of the mean (SEM). Data were analyzed using one-way analysis of variance and Fisher’s least significant difference test for multiple comparisons with SPSS 19.0 (IBM, Armonk, NY, USA). A value of P < 0.05 was considered to be statistically significant.
Results
Characterization of cultured CMECs
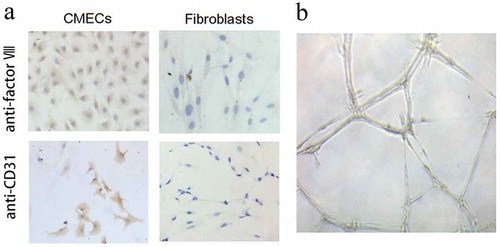
Immunocytochemistry was used to characterize the surface markers of CMECs, and the results presented in ) suggested that the CMECs expressed factor VIII and CD31, which are the main endothelial markers. The tubule formation assay further identified the lumen formation ability of CMECs ()). Taken together, the above assays suggested that the harvested cells were CMECs.
Figure 1. Characterization of cardiac microvascular endothelial cells (CMECs). (a) The characterization of CMECs was performed by immunocytochemistry, showing that CMECs factor VIII and CD31 had more than 95% of positive staining rates. Cardiac fibroblasts were used as control, in which the fibroblasts were stained as negative since these two markers (factor VIII and CD31) are endothelium-specific. (b) In vitro tubule formation assay showed that CMECs grew into a three-dimensional network of irregular tubular structure on Matrigel. All the images were taken at 100× magnification.
Integrin ανβ3 regulates LPS-induced hyperpermeability in CMECs
After CMECs were stimulated with LPS at different concentrations (10, 20, 50, 100, 1000, and 2000 ng/mL) for 6 h ()), the permeability of CMECs significantly increased compared to negative controls (without LPS stimulation), with the highest permeability being observed at 50 ng/mL of LPS. CMECs were then induced with 50 ng/mL of LPS for various durations (3, 6, 9, 12, and 18 h), showing time-dependent changes in the permeability of CMECs (from 6 h) ()). Therefore, we used 50 ng/mL of LPS and 6 h for the subsequent experiments (178 ± 15%, p < 0.01).
Figure 2. Integrin ανβ3 regulated lipopolysaccharides (LPS)-induced hyperpermeability in CMECs. (a) Changes in the permeability of CMECs monolayers after LPS stimulation with different concentration (20, 50, 100, 1000, and 2000 ng/mL) for 6 h. (b) CMECs were induced with 50 ng/mL of LPS for various time (3, 6, 9, 12, and 18 h). The absence of LPS was considered as a negative control, while the group without cells was considered as a positive control. (c) Decreased integrin ανβ3 protein levels in CMECs after LPS stimulation. After stimulation with 50 ng/mL of LPS for 6 h, the protein level of integrin ανβ3 was detected by western blotting. The top panel shows representative immunoblots showing that decreased integrin ανβ3 protein levels after LPS stimulation. β-Actin was assayed to verify the equal loading of cell lysates. The bottom panel shows the quantification of the bands by densitometry. (d) Representative immunofluorescence images for integrin ανβ3 were shown. n = 4. (100× magnificantion). (e) Integrin ανβ3 over-expression decreased LPS-stimulated CMECs permeability. Specific anti-β3 antibody and Adv-β3 were used to block and overexpress integrin-β3, respectively. CMECs permeability was measured at 6 h after stimulation with 50 ng/mL of LPS. Compared with the negative control group, *p < 0.05, **p < 0.01. Compared with the LPS group, ##p < 0.01. Each group was tested in triplicates and repeated four times.
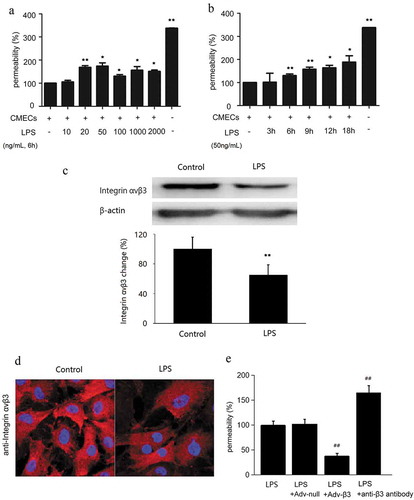
After stimulation with 50 ng/mL LPS for 6 h, the expression of integrin αvβ3 was decreased in CMECs compared with the control group ()). Furthermore, immunofluorescence showed that integrin αvβ3 expression on the cell surface was downregulated ()).
To test whether integrin αvβ3 involved in LPS-induced hyperpermeability in CMECs, we used the specific β3 integrin antibody and β3 integrin adenovirus to block and over-express integrin αvβ3. Compared with the LPS group, the permeability of the β3 integrin antibody blocking group was significantly increased (165 ± 14%, p < 0.01), while over-expression of β3 integrin decreased the cell permeability (38 ± 5%, p < 0.01). There was no significant difference between the LPS group and the null adenoviral transduction group (p > 0.05). ())
Over-expression of integrin ανβ3 relieves LPS-induced F-actin remodeling and stress fiber formation
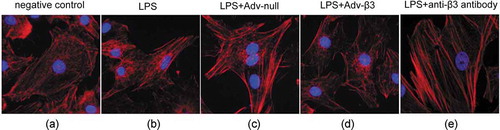
In the negative control cells, F-actin was mainly distributed in the cell periphery, the stress fibers were short and sparse, and arranged disorderly. After LPS stimulation, F-actin reconstituted to form stress fibers. The fiber filaments became thicker and longer, and arranged tightly and regularly. The upregulation of integrin β3 through adenoviral transduction inhibited F-actin remodeling and stress fiber formation. The stress fibers were short and sparse, and arranged disorderly. While, there was no significant changes of F-actin remodeling in the cells transducted with null adenoviral. Blocking of integrin β3 through antibody enhanced F-actin remodeling and stress fiber formation, indicated by thick and long stress fibers which arranged tightly and regularly. ()
Figure 3. Over-expression of integrin ανβ3 relieved LPS-induced F-actin remodeling and stress fiber formation. The altered expression of cytoskeletal protein (F-actin) was detected by staining with rhodamine-phalloidin. (a) Negative control group. (b) LPS stimulation group. (c) Null adenovirus-transduction followed by LPS stimulation. (d) ανβ3 adenovirus-transduction followed by LPS stimulation. (e) anti-ανβ3 specific antibody treatment followed by LPS stimulation. n = 5 (40× magnification).
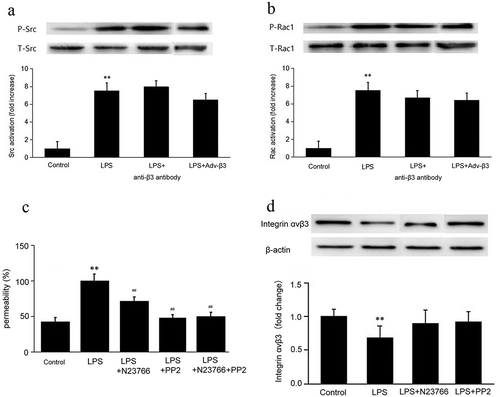
Src/Rac1 signaling is involved in integrin αvβ3-mediated inhibition of hyperpermeability in CMECs
After LPS stimulation, the activation of Rac1 and Src was increased (p < 0.01), but blocking or overexpressing integrin β3 had no effect on the activation of Rac1 and Src (p > 0.05) (,)). The CMECs monolayers pretreated with N23766, PP2, and N23766+ PP2 showed significantly decreased permeability all p < 0.01; )), but N23766 and PP2 (inhibitors of Rac1 and Src, respectively) pretreatment had no significantly effect on integrin β3 expression (p > 0.05) ()). The results indicated that ανβ3 was involved in LPS-induced hyperpermeability in a Src/Rac1-independent manner, and Src/Rac1 enhanced permeability in a ανβ3-independent manner.
Figure 4. LPS-reduced integrin αvβ3 expression was not via Src/Rac1 signaling. Specific anti-β3 antibody and Adv-β3 were used to block and overexpress integrinβ3, respectively. CMECs lysates were immunoblotted with phosphorylated (p-) or total (t-) Src (a) and Rac1 (b) and quantified. In the LPS+anti-β3 antibody group, CMECs were pretreated with anti-β3 antibody before LPS stimulation. In the LPS+Adv-β3 integrin group, CMECs were pretreated with β3 integrin adenovirus before LPS stimulation. **p < 0.01 LPS stimulation group vs. the control group. n = 3/group. (c) CMECs permeability was measured at 6 h after stimulation with 50 ng/mL of LPS by FITC-labeled albumin leakage. Permeability in the LPS group was 100%. **p < 0.01 LPS stimulation group vs. the control group. ##p < 0.01 LPS+N23766 group, LPS+PP2 group, LPS+N23766 group+PP2 group vs. LPS group. n = 3/group. (d) CMECs lysates were immunoblotted with integrin ανβ3 antibody and β-actin antibody and quantified. The N23766 and PP2 inhibitors were used to inhibit the activation of Src and Rac1, respectively. In the LPS+N23766 group, the CMECs were pretreated with N23766 for 1 h before LPS stimulation. In the LPS+PP2 group, the CMECs were pretreated with PP2 for 1 h before LPS stimulation. The top panel shows representative immunoblots showing that the integrin ανβ3 protein levels. β-Actin was assayed to verify the equal loading of cell lysates. The bottom panel shows the quantification of the bands by densitometry. **p < 0.01 LPS stimulation group vs. the control group. n = 3/group.
Discussion
The present study showed that LPS stimulation increased the CMECs permeability while it decreased integrin ανβ3 expression. Blocking integrin αvβ3 with an antibody significantly increased stress fiber formation and permeability compared with the cells in the LPS group. In addition, up-regulation of integrin ανβ3 with adenovirus decreased the LPS-induced hyperpermeability and inhibited the cytoskeletal remodeling in CMEC stimulated by LPS. Nevertheless, Src/Rac1 signaling is not involved in integrin αvβ3-mediated inhibition of hyperpermeability in CMECs.
Cytoskeletal remodeling is one of the most important mechanisms underlying vascular hyperpermeability and is closely related to microfilament structure [Citation13–Citation15]. Exposure to inflammatory factors alters the homeostasis of actin, triggering microfilament remodeling. The stress fibers mediate cytoskeletal remodeling and alter cellular morphology, resulting in the formation of intercellular gaps and transcellular holes to increase cell permeability. Integrin is the bridge to extracellular matrix and cytoskeleton. In this process, integrin is a cell adhesion receptor for many extracellular matrix proteins. Its intracellular region is connected with actin, which mediates cell signaling and cytoskeletal remodeling [Citation16,Citation17]. Su et al. in a model of sepsis and lung injury, found that S1P induces the ectopic expression of integrin ανβ3 in the cortical focal adhesion plaque to inhibit actin reconstitution and enhance vascular barrier function. The present study in CMECs indicated that blocking integrin ανβ3 significantly enhanced LPS-induced stress fibers formation, vascular permeability, and F-actin remodeling. Hence, we propose that integrin ανβ3 enhances vascular barrier function by inhibiting cytoskeletal remodeling in sepsis.
Src activation facilitates cytoskeletal remodeling, affecting intercellular and cell-matrix adhesion [Citation18]. Src kinases are involved in a variety of signaling pathways and can function as “integrators” for responding to multiple signals in LPS-induced macrophages [Citation18]. Moreover, it has been reported that Src family protein tyrosine kinases involve in the upstream signaling pathways regulate endothelial hyperpermeability via intercellular gap formation and increased transendothelial protein transport [Citation19]. Elevated activity of Src family protein tyrosine kinases can also affect endothelial permeability due to changes in gene expression [Citation20,Citation21]. Rac1 belongs to the Rho family of small GTPase. Src/Rac1 signaling pathway plays a critical role in the regulation of vascular permeability. However, the effect of Rac1 on LPS-induced vascular permeability is still controversial. On the one hand, the studies [Citation22,Citation23] found that Rac1 improved LPS-induced lung injury and reduced vascular leakage. On the other hand, Rac activation leads to hyperpermeability in LPS-stimulated endothelial cells in the study of Komarova. et al [Citation24]. Our present study showed that phosphorylation of Src and Rac were elevated in LPS-stimulated CMECs. After pretreatment with Rac1 and Src inhibitors, the LPS-stimulated CMECs showed significantly decreased permeability compared with the LPS group, suggesting that Src/Rac1 signaling enhanced permeability. Our findings were consistent with the results of Komarova.
In conclusion, integrin ανβ3 decreased LPS-induced hyperpermeability through inhibiting cytoskeletal remodeling and stress fibers formation. The underlying mechanism might be not mediated via Src/Rac1 signaling. Our study might provide new targets for the prevention and treatment of sepsis-induced cardiac injury.
Author contribution
Li Li designed the study, wrote and revised the manuscript. Zhou Rongfang conducted the experiment. Zhen Junhai and Chen Changqin contributed the data collection and data analysis. Yan Jing revised the manuscript. All of the authors read and approved the final manuscript.
Acknowledgments
We would like to show our appreciation to Dr. Chengchao Ruan for his helpful advice for this manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Leoni D, Rello J. Cardiac arrest among patients with infections: causes, clinical practice and research implications. Clin Microbiol Infect. 2016;23:730–735.
- Lagu T, Rothberg MB, Shieh MS, et al. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit Care Med. 2012;40:754–761.
- Morgan RW, Fitzgerald JC, Weiss SL, et al. Sepsis-associated in-hospital cardiac arrest: epidemiology, pathophysiology, and potential therapies. J Crit Care. 2017;40:128–135.
- Martin GS. Sepsis, severe sepsis and septic shock: changes in incidence, pathogens and outcomes. Expert Rev Anti Infect Ther. 2012;10:701–706.
- Opal SM. Endotoxins and other sepsis triggers. Contrib Nephrol. 2010;167:14–24.
- Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest. 2006;86:9–22.
- Barczyk M, Carracedo S, Gullberg D. Integrins. Cell Tissue Res. 2010;339:269–280.
- Moser M, Legate KR, Zent R, et al. The tail of integrins, talin, and kindlins. Science. 2009;324:895–899.
- Wu MH, Ustinova E, Granger HJ. Integrin binding to fibronectin and vitronectin maintains the barrier function of isolated porcine coronary venules. J Physiol. 2001;532:785–791.
- Su G, Atakilit A, Li JT, et al. Absence of integrin alphavbeta3 enhances vascular leak in mice by inhibiting endothelial cortical actin formation. Am J Respir Crit Care Med. 2012;185:58–66.
- Kilkenny C, Browne WJ, Cuthill IC, et al. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. J Pharmacol Pharmacother. 2010;1:94–99.
- Nishida M, Carley WW, Gerritsen ME, et al. Isolation and characterization of human and rat cardiac microvascular endothelial cells. Am J Physiol. 1993;264:H639–652.
- Lennon FE, Singleton PA. Hyaluronan regulation of vascular integrity. Am J Cardiovasc Dis. 2011;1:200–213.
- Castanares-Zapatero D, Bouleti C, Sommereyns C, et al. Connection between cardiac vascular permeability, myocardial edema, and inflammation during sepsis: role of the alpha1AMP-activated protein kinase isoform. Crit Care Med. 2013;41:e411–422.
- Bar-Or D. The filamentous actin cytoskeleton organization and the endothelial cell barrier. Crit Care Med. 2013;41:686–687.
- Altman SM, Dixit N, Simon SI. Detection of bidirectional signaling during integrin activation and neutrophil adhesion. Methods Mol Biol. 2014;1124:235–248.
- Morse EM, Brahme NN, Calderwood DA. Integrin cytoplasmic tail interactions. Biochemistry. 2014;53:810–820.
- Check J, Byrd CL, Menio J, et al. Src kinase participates in LPS-induced activation of NADPH oxidase. Mol Immunol. 2010;47:756–762.
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367.
- Hu G, Minshall RD. Regulation of transendothelial permeability by Src kinase. Microvasc Res. 2009;77:21–25.
- Kim MP, Park SI, Kopetz S, et al. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res. 2009;335:249–259.
- Zang G, Christoffersson G, Tian G, et al. Aberrant association between vascular endothelial growth factor receptor-2 and VE-cadherin in response to vascular endothelial growth factor-a in Shb-deficient lung endothelial cells. Cell Signal. 2013;25:85–92.
- Schlegel N, Waschke J. cAMP with other signaling cues converges on Rac1 to stabilize the endothelial barrier- a signaling pathway compromised in inflammation. Cell Tissue Res. 2014;355:587–596.
- Komarova YA, Kruse K, Mehta D, et al. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ Res. 2017;120:179–206.