
ABSTRACT
C-C motif Chemokine ligand 8 (CCL8) has been found in diseases’ pathogenesis. But its molecular mechanism in atherosclerosis (AS) remains to be elucidated. Human aortic smooth muscle cells (HASMCs) were stimulated by PDGF-BB to establish cell model. α-SMA in PDGF-BB-stimulated HASMCs was measured by immunofluorescence staining. Relative gene expressions in PDGF-BB-stimulated HASMCs were detected by quantitative real-time polymerase chain reaction and western blot. HASMCs proliferation, migration, and cell cycle were assessed by cell counting kit-8, wound-healing assay, and flow cytometry. HASMCs viability was increased after PDGF-BB stimulation, with α-SMA downregulation yet CCL8 upregulation. Silencing CCL8 inhibited PDGF-BB-stimulated HASMCs proliferation and migration, and increased cells percentage in G1 phases but decreased those in S phase. Also, silencing CCL8 decreased OPN and cyclinD1 expressions and AKT and ERK1/2 phosphorylation while increased those of α-SMA and Sm22α. However, upregulating CCL8 led to opposite effects, suggesting CCL8 could be an atherosclerosis therapeutic target.
GRAPHICAL ABSTRACT
Silencing CCL8 inhibited the proliferation and migration of PDGF-BB-stimulated human aortic smooth muscle cells.
Cardiovascular diseases (CVDs) are defined as a series of diseases which are mainly involved in heart vessels [Citation1]. Atherosclerosis, a common type of CVD, refers to the process of accumulation of fatty and/or fibrous material in the innermost layer of arteries, which has caused much morbidity and mortality around the world [Citation2]. Atherosclerosis is a complex and multi-step process which prevents the blood flow to the heart, brain, or lower extremities thus cause the onset of many other CVDs [Citation3]. It has been discussed by many scholars both home and abroad that some molecular mechanisms are involved in the onset of the disease [Citation4]. Many evidences from scientific researches have shown that the abnormal proliferation and migration of vascular smooth muscle cells (VSMCs) may be a crucial element which may induce to the atherosclerosis development and progression [Citation5,Citation6].
α-SMA, an isoform of vascular smooth muscle actin, is encoded by actin α 2, smooth muscle and is mainly expressed in VSMCs, which induces the motility and contraction of vascular cells [Citation7]. Sm22α, an actin-binding protein, is shown to enhance VSMCs contractility and mobility, and it is discovered that Sm22α activation contributes to the balance of VSMCs differentiated phenotype [Citation7]. OPN is seen as a biomarker of VSMC phenotypic switch and plays an active role in the development of vascular remodeling diseases like atherosclerosis [Citation8]. CyclinD1 is a protein for progression through the G1 phase of cell cycle and it is proposed to act as an active switch in regulating the continued cell cycle progression [Citation9,Citation10].
At present, more and more experimental reports indicated that chemokine played a significant role in atherosclerosis development and progression [Citation11,Citation12]. Chemokine is defined as a small yet highly conserved protein family which is found involved in many biological process [Citation13]. In general, chemokine can be further categorized into several subclasses according to their sequential positioning of those highly conserved cysteine residues: CC chemokines, CXC chemokines, C chemokines, and CX3 C chemokines [Citation14]. CC chemokines, a subfamily of chemokines, are found implicated in disease pathologies in which angiogenesis driven by inflammation played a significant role and many of them have been recognized in atherosclerotic plaques [Citation15].
CC chemokine subfamily was defined according to the arrangement of the first two of four invariant cysteine residues which could be found in all chemokines [Citation16]. Its function was shown to mediate and direct the trafficking and migration of monocytes as well as lymphocytes [Citation16]. Chemokine C-C motif ligand 8 (CCL8), which is also known as monocyte chemoattractant protein-2 (MCP-2), is a small cytokine member which belongs to the CC chemokine subfamily [Citation17]. It has been discovered that CCL8 could interact with multiple cellular chemokine receptors [Citation18], such as CCR3 [Citation19], and could attract and activate human leukocytes [Citation20]. In a scientific investigation in the molecular mechanism of CCL8, it has shown that CCL8 may play an important role in atherosclerosis and may be used as an indicator for coronary artery diseases (CADs) [Citation21]. However, detailed discussion on the effects of upregulating or silencing CCL8 in AS progression was inadequate. Therefore, in this study, we measured the effects of upregulating or silencing CCL8 on PDGF-BB-induced HASMCs, with the hope to find a potential therapeutic curing method for AS.
Materials and methods
Cell culture and exposure to platelet-derived growth factor BB (PDGF-BB)
In this study, human aortic smooth muscle cells (HASMCs) were bought from American Type Culture Collection (PCS-100-012; ATCC; Rockville, MD, USA) (https://www.atcc.org/) and were cultured in smooth muscle cell medium (SMCM, 3 H Biomedical, Uppsala, Sweden) which is supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA), 100 U/mL penicillin (Invitrogen, USA) and 100 mg/mL streptomycin (Invitrogen, USA) in a humidified incubator at 37°C with 5% CO2. HASMCs were seeded in a six-well plate at a density of 1 × 105 cells/well. Then, they were exposed to 20 ng/mL PDGF-BB (Peprrotech, USA) for 48 h to establish the AS cell model.
Cell Transfection
For transfection, HASMCs were seeded in a six-well plate at a density of 1.5 × 106 cells/well. One hundred nM CCL8 siRNA (siCCL8) or control siRNA were synthesized by Invitrogen. The overexpression of CCL8 was made with the pcDNA3.1 plasmid (Thermo Fisher Scientific, Waltham, MA, USA). The cells were divided into control (untreated cells), PDGF-BB, NC+PDGF-BB, CCL8+ PDGF-BB, and siCCL8+ PDGF-BB group. The transfection was later performed with Lipofectamine 2000 transfection reagent (Invitrogen, USA) according to the protocol of manufacturer. The cells were harvested after 48 h of transfection for the following experiments. Then, successful transfections were determined by RT-qPCR and Western blot assay after cells were incubated for 48 h.
Immunofluorescence staining and analysis
HASMCs were seeded in 24-well plates, after 20 ng/mL PDGF-BB treatment, cells were fixed with freshly prepared 4% paraformaldehyde for 15 min, followed by permeabilization with 0.2% Triton X-100 (Sigma-Aldrich, Inc., St Louis, MO, USA) in PBS for 20 min at room temperature. Having been blocked in 5% bovine serum albumin (BSA) for 30 min, the cells were incubated with anti-α-SMA (rabbit, ab32575, 1:20, Abcam, Cambridge, UK) at 4°C overnight. Then, the cells were incubated with secondary antibody (goat, ab205718, 1:2000, Abcam, UK) for 1 h at room temperature after being washed three times with PBS. Finally, the nuclei were stained by 4ʹ, 6-diamidino-2-phenylindole (DAPI; D1306; Thermo Fisher Scientific, USA) for 10 min, and then the cells were washed three times with PBS in the dark. Immunofluorescence in the cells was later analyzed under a laser confocal microscope (LSM980, Carl Zeiss Microscopy, Oberkochen, Germany) following the manufacturer’s manuals.
Cell Counting Kit-8 (CCK-8) assay
To find out the effect of CCL8 silencing or overexpression on the proliferation of HAMSCs stimulated by PDGF-BB, after different treatment, cells were cultured in an incubator at 37°C with 5% CO2 for 24 and 48 h. Then, 10 μL CCK-8 reagent (Dojindo, Tokyo, Japan) was then introduced to detect the cell viability. After the incubation at 37°C for 1 h, the OD values of each sample cells were measured and determined by microplate reader at an absorbance of 450 nm (Model 680, Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Wound-healing assay
In vitro HASMCs migration was measured by wound-healing assay. After transfecting for 48 h, HASMCs were seeded in six-well culture plates at a density of 1 × 105 cells/well. A sterile pipette tip was used to create a straight wound in the middle of the culture after cells merged into the monolayer. Then, the cells were washed by phosphate-buffered saline (PBS) for 2 times to smooth the edge and remove the floating cells, followed by being incubated in an incubator at 37°C with 5% CO2. Cell images at 0 and 48 h were captured and analyzed under a compound microscope system (Olympus Corporation, Tokyo, Japan). The cells migration was under the measurement of Image-Pro Plus Analysis Software (Version 6.0, Media Cybernetics Company, USA).
Flow Cytometry
The cell cycle analysis kit (K920, BioVision, Inc., CA, USA) was applied to perform cell cycle analysis. After transfection for 48 h, for increasing cell membrane penetrability, 70% cool ethanol was added into 1 × 105 cells at 4°C overnight. Then, RNase (Thermo Fisher Scientific, USA) was applied to treat cells at 37°C for 20 min. After staining with propidium iodide (PI, Sangon Biotech, Shanghai, China) in the dark at room temperature, cell cycle analysis was performed with a flow cytometer (NovoCyte 2090 V; ACEA Biosciences, Inc., San Diego, CA, USA).
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
According to the protocol of the manufacturer, total RNA from the cells was extracted with Trizol reagent (Invitrogen, Madison, WI, USA). Concentration of total RNA was detected and quantified by a biological spectrometer (Nano Drop 2000, Thermo Fisher, Waltham, MA, USA). The cDNA was synthesized from total RNA (1 μg) by a First-strand cDNA Synthesis Kit (NP100041, Origene, Rockville, MD, USA) under the instructions of the manufacturer. QRT-PCR experiment was performed with SYBR PremixEx Taq II kit (RR820 L, TaKaRa, Japan) in Touch real-time PCR Detection system (CFX384, Bio-Rad, USA). The experiment was conducted in the following conditions: 95°C for 5 min, then 40 cycles of 94°C for 15 s, 55°C for 20 s and 72°C for 20 s, followed by 72°C for 7 min. Primer sequences for qRT-PCR in the experiments are shown in . GAPDH was used as internal reference, and the relative genes expressions were analyzed and quantified by 2−ΔΔCT calculation method [Citation22].
Table 1. Primers of qRT-PCR.
Western blotting
After transfection for 48 h, HASMCs were collected and the protein was extracted with RIPA lysis buffer (Thermo Fisher Scientific, USA). BCA protein kit (Sigma-Aldrich, USA) was chosen to test protein concentration and 20 μg of sample protein lysates were then electrophoresed by 12% SDS-PAGE (Beyotime, Shanghai, China; P0012A), followed by the process of transferring into polyvinylidene fluoride membrane (PVDF; Beyotime; FFP28). The membrane was subsequently blocked with 5% nonfat milk for 2 h and incubated in the primary antibodies which contains anti-α-SMA (rabbit, ab32575, 1:1000, Abcam, Cambridge, UK), 1 µg/mL anti-Sm22α (rabbit, ab14106, Abcam, UK), anti-OPN (rabbit, ab214050, 1:1000, Abcam, UK), anti-cyclinD1 (rabbit, ab16663, 1:200, Abcam, UK), anti-p-ERK1/2 (rabbit, ab214362, 1:1000, Abcam, UK), anti-ERK1/2 (rabbit, ab184699, 1:10,000, Abcam, UK), anti-p-AKT (rabbit, ab38449, 1:1000, Abcam, UK), 1 µg/mL anti-AKT (rabbit, ab64148, Abcam, UK), 0.2 µg/mL anti-CCL8 (rabbit, ab39625, Abcam, UK) and anti-GAPDH (rabbit, ab181602, 1:10,000, Abcam, UK) at 4°C overnight. GAPDH was used as the internal reference. Then, the membrane was incubated in the secondary horseradish peroxidase (HRP)-combined antibody: goat anti-rabbit IgG H&L (HRP) (goat; 1:2000, Abcam, UK) for 1 h and washed with tris-buffer saline tween (TBST) for three times. The protein band collected from the samples was under the analysis of enhanced chemiluminescence (ECL) kit (BioLegend, San Diego, CA, USA) and the gray values of the strips were further gathered and calculated by ImageJ (version 5.0; Bio-Rad, Hercules, CA, USA).
Statistical analysis
All data from the experiments were quantified as mean ± standard deviation (SD). Statistical analysis was conducted by SPSS19.0 software (IBM Corporation, Armonk, NY, USA). Statistical comparisons were made by Student’s t-test and one-way ANOVA followed by Dunnett’s post hoc test. P-values below 0.05 were considered statistically significant.
Results
Effects of PDGF-BB on cell viability, CCL8, and α-SMA expressions in HASMCs
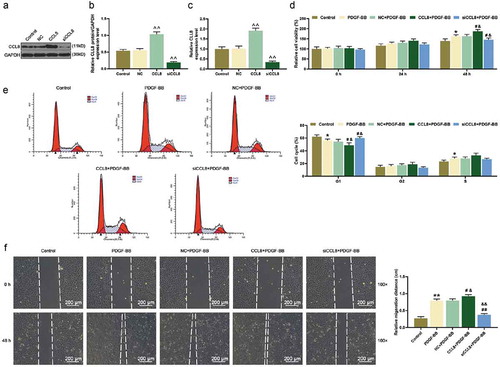
In this study, in order to confirm whether the AS cells model was established successfully, the immunofluorescence staining and western blot were applied. We used 20 ng/mL PDGF-BB to stimulate HASMCs and it was discovered that the viability of HASMCs was increased significantly at 48 h ()) while the number of α-SMA in HASMCs was seen decreased in immunofluorescence imaging at 24 h ()). Furthermore, experimental data from western blot and qRT-PCR unveiled that the protein and mRNA expression of α-SMA in HASMCs were downregulated but the protein and mRNA expression level of CCL8 were upregulated after PDGF-BB stimulation ().
Figure 1. Cell viability, CCL8, and α-SMA expression in HASMCs under PDGF-BB stimulation were detected. (a) Cell Counting Kit-8 (CCK-8) assay was used to detect the cell viability after PDGF-BB stimulation at 0, 24, and 48 h. *P < 0.05, vs. Control. (b) Immunofluorescence staining was used to detect α-SMA in HASMCs stimulated by PDGF-BB at 0 and 24 h. magnification: 400 ×. (c-d) The protein expression level of α-SMA and CCL8 in HASMCs after PDGF-BB stimulation was detected by western blot. **P < 0.01, vs. Control. (e) The mRNA expression level of α-SMA and CCL8 in HASMCs after PDGF-BB stimulation was detected by qRT-PCR. **P < 0.01, vs. Control. HASMCs: human aortic smooth muscle cells; PDGF-BB: platelet-derived growth factor BB; CCL8: C-C motif Chemokine ligand 8.
Effects of siCCL8 and overexpressed CCL8 on cells proliferation in HASMCs stimulated by PDGF-BB
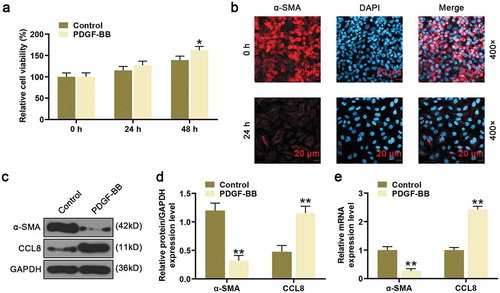
To test the effects of CCL8 on HASMCs proliferation, CCL8 was down-regulated or up-regulated by transfection with siCCL8 or CCL8. It was found that the CCL8 expression level in HASMCs was increased after transfected with CCL8 but silencing CCL8 in HASMCs greatly decreased CCL8 expression compared with NC group (); in addition, PDGF-BB increased the expression of CCL8, the expression of CCL8 was higher in CCL8+ PDGF-BB group than that in PDGF-BB group and CCL8 group, and CCL8 expression was higher in siCCL8+ PDGF-BB group than that in siCCL8 group (). After PDGF-BB stimulation, the cell viability was increased ()). In addition, after upregulating CCL8 in PDGF-BB-stimulated HASMCs, the cell viability was raised in CCL8+ PDGF-BB group compared with PDGF-BB group and NC+PDGF-BB group, while silencing CCL8 inhibited cell viability comparison with PDGF-BB group and NC+PDGF-BB group ()). Further to this, the results from cell cycle analysis showed that after PDGF-BB stimulation, the percentage of HASMCs at G1 phase was reduced while the percentage at S phase was going up. Also, after upregulating CCL8 in PDGF-BB-induced cells, the percentage of HASMCs at G1 phase was reduced. However, after silencing CCL8 in the cells, the percentage of HASMCs in siCCL8+ PDGF-BB group at G1 phase was significantly increased, which induced to a cycle arrest ()).
Figure 2. Effects of CCL8 on proliferation, migration, and cell cycle in HASMCs stimulated by PDGF-BB. (a-b) CCL8 protein level after upregulating or silencing CCL8 in HASMCs or PDGF-BB-induced HASMCs was measured by western blot. **P < 0.01 and ***P < 0.001, vs. Control; ##P < 0.01, vs. PDGF-BB; &&P < 0.01, vs. NC+PDGF-BB; ^^P < 0.01, vs. CCL8+ PDGF-BB; §§P < 0.01 and §§§P < 0.001, vs. siCCL8+ PDGF-BB. (c) CCL8 mRNA expression level after upregulating or silencing CCL8 in HASMCs or PDGF-BB-induced HASMCs was measured by qRT-PCR. **P < 0.01 and ***P < 0.001, vs. Control; ##P < 0.01, vs. PDGF-BB; &&P < 0.01, vs. NC+PDGF-BB; ^^P < 0.01, vs. CCL8+ PDGF-BB; §§P < 0.01 and §§§P < 0.001, vs. siCCL8+ PDGF-BB. (d) The viability of PDGF-BB-induced HASMCs after upregulating or silencing CCL8 was detected by CCK-8 assay at 0, 24, and 48 h. *P < 0.05, vs. Control; #P < 0.05, vs. PDGF-BB; &P < 0.05, vs. NC+PDGF-BB. (e) Cell cycle analysis in each group was detected using flow cytometry. *P < 0.05, vs. Control; #P < 0.05, vs. PDGF-BB; &P < 0.05, vs. NC+PDGF-BB. (f) PDGF-BB-induced HASMCs migration rates after upregulating or silencing CCL8 was measured by wound-healing assay. **P < 0.01, vs. Control; &P < 0.05 and &&P < 0.01, vs. NC+PDGF-BB; #P < 0.05, and ##P < 0.01, vs. PDGF-BB. HASMCs: human aortic smooth muscle cells; PDGF-BB: platelet-derived growth factor BB; CCL8: C-C motif Chemokine ligand 8; NC: negative control.
Effects of siCCL8 and overexpressed CCL8 on cell migration in HASMCs stimulated by PDGF-BB
In this stage, we observed cells migration by wound-healing assay. From the results of the study, the relative migration distance in cells after PDGF-BB stimulation was significantly increased after 48 h compared with control group ()). Moreover, after upregulating CCL8 in the PDGF-BB-stimulated cells, the relative migration distance was also found increased in comparison with PDGF-BB group and NC+PDGF-BB group ()). However, after silencing CCL8 in the cells, the relative migration distance was reduced compared with PDGF-BB group and NC+PDGF-BB group ()).
Effects of siCCL8 and overexpressed CCL8 on α-SMA, Sm22α, OPN, and cyclinD1 expressions in HASMCs stimulated by PDGF-BB
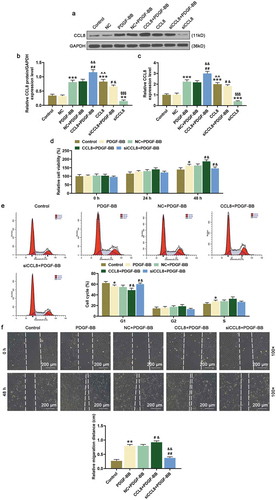
In order to further uncover the molecular mechanism of CCL8 on HASMCs, we detected the protein and mRNA expression of α-SMA, Sm22α, OPN, and cyclinD1 in HASMCs stimulated by PDGF-BB after upregulating or silencing CCL8. After PDGF-BB stimulation, α-SMA and Sm22α protein and mRNA expressions in HASMCs were decreased, while the protein and mRNA expressions of OPN and cyclinD1 were increased (). In addition, after upregulating CCL8, both protein and mRNA expressions of α-SMA and Sm22α were further decreased, while those of OPN and cyclinD1 were increased, compared with PDGF-BB and NC+PDGF-BB group (). However, silencing CCL8 upregulated the protein and mRNA expressions of α-SMA and Sm22α yet downregulated those of OPN and cyclinD1 ().
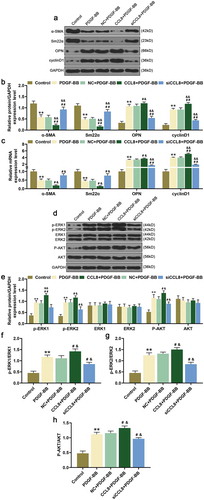
Figure 3. Effects of CCL8 on the expression of α-SMA, Sm22α, OPN, cyclinD1, p-ERK1/2, and p-AKT were detected by western blot and qRT-PCR. (a-b) Western blot assay was used to measure the protein expression level of α-SMA, Sm22α, OPN, and cyclinD1 in HASMCs stimulated by PDGF-BB after upregulating or silencing CCL8. **P < 0.01, vs. Control; #P < 0.05 and ##P < 0.01, vs. PDGF-BB; &P < 0.05 and &&P < 0.01 vs. NC+PDGF-BB. (c) The relative mRNA expressions level of α-SMA, Sm22α, OPN, and cyclinD1 in HASMCs stimulated by PDGF-BB after upregulating or silencing CCL8 was measured by qRT-PCR. **P < 0.01, vs. Control; &P < 0.05 and &&P < 0.01, vs. NC+PDGF-BB; #P < 0.05 and ##P < 0.01, vs. PDGF-BB. (d-e) Western blot assay was adopted to measure the protein expression levels of p-ERK1/2, ERK1/2, p-AKT as well as AKT in HASMCs stimulated by PDGF-BB after CCL8 upregulation or silencing. **P < 0.01, vs. Control; &P < 0.05 and &&P < 0.01, vs. NC+PDGF-BB; #P < 0.05 and ##P < 0.01, vs. PDGF-BB. (f) The phosphorylation level of ERK1 in HASMCs stimulated by PDGF-BB after upregulating or silencing CCL8 was detected by western blot. **P < 0.01, vs. Control; #P < 0.05, vs. PDGF-BB; &P < 0.05, vs. NC+PDGF-BB. (g) The phosphorylation level of ERK2 in HASMCs stimulated by PDGF-BB after upregulating or silencing CCL8 was determined by western blot. **P < 0.01, vs. Control; #P < 0.05, vs. PDGF-BB; &P < 0.05, vs. NC+PDGF-BB. (h) The phosphorylation level of AKT in HASMCs stimulated by PDGF-BB after upregulating or silencing CCL8 was quantified by western blot. **P < 0.01, vs. Control; #P < 0.05, vs. PDGF-BB; &P < 0.05, vs. NC+PDGF-BB. HASMCs: human aortic smooth muscle cells; PDGF-BB: platelet-derived growth factor BB; CCL8: C-C motif Chemokine ligand 8; NC: negative control.
Effects of siCCL8 and overexpressed CCL8 on the phosphorylation of ERK1/2 and AKT in HASMCs stimulated by PDGF-BB
In this section, we detected the effect of CCL8 on the phosphorylation level of ERK1/2 and AKT in HASMCs. After PDGF-BB stimulation, the protein expressions of p-ERK1/2 and p-AKT in HASMCs were increased compared with control group (). Meanwhile, after upregulating CCL8 in HASMCs stimulated by PDGF-BB, the protein expression of p-ERK1/2 and p-AKT was also raised compared with PDGF-BB group and NC+PDGF-BB group (). However, the protein expression of p-ERK1/2 and p-AKT was decreased after silencing CCL8 in HASMCs stimulated by PDGF-BB in comparison with PDGF-BB group and NC+PDGF-BB group ().
Furthermore, we verified the phosphorylation of ERK1/2 and AKT in HASMCs. In this section, we uncovered that after PDGF-BB stimulation, the phosphorylation of ERK1/2 and AKT in HASMCs was evidently upregulated compared with Control group (). The same result was discovered after upregulating CCL8, which suggested that CCL8 upregulation could promote the phosphorylation of ERK1/2 and AKT in HASMCs. On the contrary, after silencing CCL8, the phosphorylation of ERK1/2 and AKT in HASMCs was dropped (). In conclusion, regulating CCL8 in HASMCs stimulated by PDGF-BB may affect the phosphorylation level of ERK1/2 and AKT.
Discussion
In atherosclerosis pathology progression, vascular injuries lead to the unleash of multiple growth factors in VSMCs [Citation23]. And platelet-derived growth factor (PDGF) is found to regulate blood vessel formation [Citation24]. PDGF family consists of five kinds of homodimers, which includes PDGF-AA, PDGF-BB, PDGF-AB, PDGF-CC as well as PDGF-DD [Citation25]. Among them, PDGF-BB is said to be one of the most potential stimulants for phenotypical switching of VSMCs from a contractile phenotype to a synthetic one [Citation26]. Consequently, PDGF-BB is often adopted as a stimulant to induce the phenotypical switching of VSMCs [Citation27]. In our study, PDGF-BB was used to stimulate HASMCs as well. The results demonstrated that PDGF-BB promoted HASMCs proliferation and migration.
It has been reported that circulating chemokines (such as CCL8) accurately identify individuals with clinically significant atherosclerotic heart disease, CCL8 is highly expressed in atherosclerosis heart disease [Citation28]. Several studies on CCL8 demonstrated that CCL8 expression was increased in glioma cells [Citation29]. CCL8 was discovered to be targeted by miR-181 and downregulating CCL8 could inhibit glioma cells proliferation and migration [Citation29]. Moreover, CCL8 was found to drive the dissemination of breast cancer cells [Citation30]. Yet in our present studies, CCL8 was seen to be upregulated in HASMCs which were under the stimulation of PDGF-BB. Upregulating CCL8 expression promoted the HASMCs stimulated by PDGF-BB proliferation and migration as well as accelerated the cell cycle and the phenotypic switch by regulating the expression of key molecules in HASMCs, and silencing CCL8 had opposite effect. It has been reported that silencing of CCL8 in tumor-supportive fibroblasts reduces their ability to promote tumorigenicity [Citation31].
VSMCs in normal arteries are often quiescent, showing a contractile phenotype which is found connected to the stable production of some specific proteins such as α-SMA, Sm22α, which are seen as some of VSMCs markers [Citation32]. Further to this, it has been observed in our studies that VSMCs under atherosclerosis could downregulate the expression of those markers and switch to a proinflammatory phenotype with a strong capability of proliferation and migration. This switch of VSMCs has been thought to be of great significance to atherosclerosis progression [Citation33]. It was found in Zhang’s studies that cyclinD1 overexpression partially reversed the inhibitory effects of miR-365 on VSMCs proliferation [Citation10]. In our studies, we first discovered that after upregulating CCL8 in HASMCs, the protein expressions of α-SMA and Sm22α were decreased while those of OPN and cyclinD1 were increased, while silencing CCL8 promoted the expression of α-SMA and Sm22α, and inhibited OPN and cyclinD1 expressions, showing that CCL8 upregulation may promote the proliferation and migration of HASMCs as well as accelerate the cell cycle and the phenotypic switch, thus lead to the onset of atherosclerosis. However, this study also has certain limitations. We only detected CCL8 in PDGF-BB-stimulated HASMCs, and it is possible that PDGF-BB may also lead to the expression of other inflammatory cytokines (such as TNF-α, IL-1β, or IL-8), which needs further analysis.
In conclusion, CCL8 was found upregulated in HASMCs stimulated by PDGF-BB. Upregulating CCL8 was shown to have inducing effects on HASMCs stimulated by PDGF-BB proliferation, migration, and cell cycle, as well as the phenotypic switch of HASMCs. However, silencing CCL8 inhibited the proliferation, migration, and cell cycle, as well as the phenotypic switch of PDGF-BB-stimulated HASMCs. In addition, the phosphorylation of ERK1/2 and AKT in HASMCs stimulated by PDGF-BB was blocked after silencing CCL8. The discoveries from our experiments suggested that silencing CCL8 in HASMCs might be adopted as an effective clinical treatment for atherosclerosis.
Authors’ contributions
Substantial contributions to conception and design: SD
Data acquisition, data analysis, and interpretation: JZ, ZX
Drafting the article or critically revising it for important intellectual content: SD
Final approval of the version to be published: All authors
Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of the work are appropriately investigated and resolved: All authors
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
No animals are involved in this research.
Disclosure statement
The authors declare no conflicts of interest.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- Libby P, Bornfeldt KE, Tall AR. Atherosclerosis: successes, surprises, and future challenges. Circ Res. 2016 Feb 19;118(4):531–534. PubMed PMID: 26892955; PubMed Central PMCID: PMCPMC4762065. eng.
- Libby P, Buring JE, Badimon L, et al. Atherosclerosis. Nat Rev Dis Primers. 2019 Aug 16;5(1):56. PubMed PMID: 31420554; eng.
- Zhang MJ, Zhou Y, Chen L, et al. SIRT1 improves VSMC functions in atherosclerosis. Prog Biophys Mol Biol. 2016 May;121(1):11–15. PubMed PMID: 27080738; eng.
- Kwan P, Alexis D, Tredget E. Molecular and cellular basis of hypertrophic scarring. Elsevier Inc. 2018. p. 455–465.e4.
- Lim S, Park S. Role of vascular smooth muscle cell in the inflammation of atherosclerosis. BMB Rep. 2014;47(1):1–7. PubMed PMID: 24388105; eng.
- Basatemur GL, Jørgensen HF, Clarke MCH, et al. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. 2019 June 26;16(12):727–744.
- Yuan SM. alpha-smooth muscle actin and ACTA2 gene expressions in vasculopathies. Braz J Cardiovasc Surg. 2015 Nov–Dec;30(6):644–649. PubMed PMID: 26934405; PubMed Central PMCID: PMCPMC4762557. eng.
- Lee SJ, Baek SE, Jang MA, et al. Osteopontin plays a key role in vascular smooth muscle cell proliferation via EGFR-mediated activation of AP-1 and C/EBPbeta pathways. Pharmacol Res. 2016 Jun;108:1–8. PubMed PMID: 27089830; eng.
- Stacey DW. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr Opin Cell Biol. 2003 Apr;15(2):158–163. PubMed PMID: 12648671; eng.
- Zhang P, Zheng C, Ye H, et al. MicroRNA-365 inhibits vascular smooth muscle cell proliferation through targeting cyclin D1. Int J Med Sci. 2014;11(8):765–770. PubMed PMID: 24936138; eng.
- Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000 Feb;12(2):121–127. PubMed PMID: 10714678; eng.
- Apostolakis S, Spandidos D. Chemokines and atherosclerosis: focus on the CX3CL1/CX3CR1 pathway. Acta Pharmacol Sin. 2013 Oct;34(10):1251–1256. PubMed PMID: 23974513; PubMed Central PMCID: PMCPMC4002164. eng.
- Miller MC, Mayo KH. Chemokines from a structural perspective. Int J Mol Sci. 2017 Oct 2;18(10). PubMed PMID: 28974038; PubMed Central PMCID: PMCPMC5666770. eng. DOI:10.3390/ijms18102088
- Ramji DP, Davies TS. Cytokines in atherosclerosis: key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev. 2015;26(6):673–685. PubMed PMID: 26005197; eng.
- Ridiandries A, Tan JT, Bursill CA. The role of CC-chemokines in the regulation of angiogenesis. Int J Mol Sci. 2016 Nov 8;17(11):1856. PubMed PMID: 27834814; PubMed Central PMCID: PMCPMC5133856. eng.
- Santoni M, Bracarda S, Nabissi M, et al. CXC and CC chemokines as angiogenic modulators in nonhaematological tumors. Biomed Res Int. 2014;2014:768758. PubMed PMID: 24971349; PubMed Central PMCID: PMCPMC4058128. eng.
- Maddaluno M, Di Lauro M, Di Pascale A, et al. Monocyte chemotactic protein-3 induces human coronary smooth muscle cell proliferation. Atherosclerosis. 2011 Jul;217(1):113–119. PubMed PMID: 21536288; eng.
- Blaszczyk J, Coillie EV, Proost P, et al. Complete crystal structure of monocyte chemotactic protein-2, a CC chemokine that interacts with multiple receptors. Biochemistry. 2000 Nov 21;39(46):14075–14081. PubMed PMID: 11087354; eng.
- Ge B, Li J, Wei Z, et al. Functional expression of CCL8 and its interaction with chemokine receptor CCR3. BMC Immunol. 2017;18(1):54. PubMed PMID: 29281969; eng.
- Zhou J, Zheng S, Liu T, et al. MCP2 activates NF-kappaB signaling pathway promoting the migration and invasion of ESCC cells. Cell Biol Int. 2018 Mar;42(3):365–372. PubMed PMID: 29148603; eng.
- Yu X, Guan W, Zhang Y, et al. Large-scale gene analysis of rabbit atherosclerosis to discover new biomarkers for coronary artery disease. Open Biol. 2019 Jan 31;9(1):180238. PubMed PMID: 30958112; PubMed Central PMCID: PMCPMC6367139. eng.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001 Dec;25(4):402–408. PubMed PMID: 11846609; eng.
- van Hout GPJ, Bosch L. The inflammasomes in cardiovascular disease. Experientia Supplementum (2012). 2018;108:9–40. PubMed PMID: 30536166; eng.
- Dong X, Hu H, Fang Z, et al. CTRP6 inhibits PDGF-BB-induced vascular smooth muscle cell proliferation and migration. Biomed Pharmacothe. 2018 Jul;103:844–850. PubMed PMID: 29710500; eng.
- Bartoschek M, Pietras K. PDGF family function and prognostic value in tumor biology. Biochem Biophys Res Commun. 2018 Sep 5;503(2):984–990. PubMed PMID: 29932922; eng.
- Lu QB, Wan MY, Wang PY, et al. Chicoric acid prevents PDGF-BB-induced VSMC dedifferentiation, proliferation and migration by suppressing ROS/NFkappaB/mTOR/P70S6K signaling cascade. Redox Biol. 2018 Apr;14:656–668. PubMed PMID: 29175753; PubMed Central PMCID: PMCPMC5716955. eng.
- Chen S, Liu B, Kong D, et al. Atorvastatin calcium inhibits phenotypic modulation of PDGF-BB-induced VSMCs via down-regulation the Akt signaling pathway. PloS One. 2015;10(4):e0122577. PubMed PMID: 25874930; PubMed Central PMCID: PMCPMC4398430. eng.
- Ardigo D, Assimes TL, Fortmann SP, et al. Circulating chemokines accurately identify individuals with clinically significant atherosclerotic heart disease. Physiol Genomics. 2007 Nov 14;31(3):402–409. PubMed PMID: 17698927; eng.
- Zhai F, Chen X, He Q, et al. MicroRNA-181 inhibits glioblastoma cell growth by directly targeting CCL8. Oncol Lett. 2019;18(2):1922–1930. PubMed PMID: 31423262; eng.
- Farmaki E, Chatzistamou I, Kaza V, et al. A CCL8 gradient drives breast cancer cell dissemination. Oncogene. 2016 Dec 8;35(49):6309–6318. PubMed PMID: 27181207; PubMed Central PMCID: PMCPMC5112152. eng.
- Tank J, Lindner D, Wang X, et al. Single-target RNA interference for the blockade of multiple interacting proinflammatory and profibrotic pathways in cardiac fibroblasts. J Mol Cell Cardiol. 2014 Jan;66:141–156. PubMed PMID: 24239602; eng.
- Zhang MJ, Zhou Y, Chen L, et al. An overview of potential molecular mechanisms involved in VSMC phenotypic modulation. Histochem Cell Biol. 2016 Feb;145(2):119–130. 10.1007/s00418-015-1386-3. PubMed PMID: 26708152; eng.
- Davies M, Harman J, Yu H, et al. The epigenetic phenotypic switch of vascular smooth muscle cells involved in atherosclerosis. Lancet. 2013;381:S34.