
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Molecular chaperones play vital roles in various physiological reactions by regulating the folding and assembly of biomacromolecules. We have demonstrated that cationic comb-type copolymers exhibit chaperone activity for anionic biomolecules including DNA and ionic peptide via the formation of soluble interpolyelectrolyte complexes. The development of smart artificial chaperones that can be spatiotemporally controlled by a remotely guided signal would expand the functions of artificial chaperones. Herein, to enable photocontrol of chaperone activity, a cationic comb-type copolymer bearing malachite green as a photoresponsive unit was designed. We first prepared a series of carboxylic acid derivatives of malachite green identified a derivative that could be quickly and quantitatively converted to the cationic form from the nonionic form by photoirradiation. This derivative was conjugated to the cationic comb-type copolymer, poly(allylamine)-graft-poly(ethylene glycol) through a condensation reaction. Upon photoirradiation, the copolymer bearing 9 mol% malachite green enhanced the membrane disruptive activity of acidic peptide E5 and induced morphological changes in liposomes. This demonstration of photoresponsive activation of chaperoning activity of a copolymer suggests that the installation of carboxyl derivatives of malachite green will impart photoresponsiveness to various materials including biopolymers.
1. Introduction
Biomacromolecules can form a variety of higher-order structures stabilized by noncovalent interactions such as hydrogen bonds, hydrophobic interactions, and π–π stacking interactions. Misfolding and irreversible aggregation of proteins induced by stresses such as high temperature, high protein concentration, and high or low pH can result in the accumulation of misfolded proteins, and protein misfolding is thought to underlie the pathologies of serious diseases such as Alzheimer’s and Parkinson’s diseases [Citation1–3]. The correct folding and the refolding of biomolecules are catalyzed by molecular chaperones, a family of cellular proteins. For example, the GroEL/GroES system in Escherichia coli uses energy from the hydrolysis of ATP to catalyze the refolding of denatured proteins [Citation4,Citation5]. Artificial materials that possess functions of natural molecular chaperones have been identified. For example, Akiyoshi et al. demonstrated that nanogels composed of hydrophobized pullulan act as artificial chaperones. The nanogels spontaneously capture denatured proteins via hydrophobic interactions. Upon the addition of cyclodextrin, the captured proteins are released from the nanogel in native form [Citation6]. Lower critical solution temperature-type thermoresponsive polymers such as poly(N-isopropylacrylamide), which undergo a phase transition from a hydrophilic state to a hydrophobic state upon heating, have also been shown to induce protein refolding [Citation7,Citation8].
Our group has designed cationic comb-type copolymers as artificial chaperones for nucleic acids. The cationic copolymers, which consist of polycationic backbones and hydrophilic graft chains, form soluble interpolyelectrolyte complexes with negatively charged DNA strands. The copolymers promote DNA hybridization by relieving the electrostatic repulsions between DNA strands [Citation9,Citation10]. These copolymers markedly accelerate DNA strand displacement reactions [Citation11,Citation12], and this chaperoning activity enhances activities of DNA molecular devices such as DNA computers [Citation13], nanomachines [Citation14], hybridization chain reactions [Citation15], and DNA enzymes [Citation16–19]. Recently, we found that the copolymers chaperone an amphiphilic and anionic peptide, E5, derived from a membrane protein of influenza virus into active conformation [Citation20–22]. The copolymers significantly enhanced the membrane-disruptive activity of E5 [Citation23], and in the presence of the copolymer but not its absence, E5 triggered transformation of lipid vesicles into lipid sheets in all-or-none manner under physiological conditions [Citation24].
Light-mediated regulation of the chaperone activity that permits spatiotemporal targeting capability and low invasiveness should expand the range of biotechnological applications of the copolymers. Photochromic molecules have been widely employed for the preparation of photoresponsive materials. For example, spiropyran derivatives are isomerized from the hydrophobic spiro-form to the hydrophilic mero-form upon photostimulation [Citation25]. Spiropyran-based smart materials are used for photoregulation of micellar aggregation [Citation26,Citation27], cell adhesion [Citation28,Citation29], and DNA release [Citation30]. Akiyoshi et al. modified pullulan nanogels with spiropyran derivatives to allow photoresponsive capture and release of proteins [Citation31].
Herein, we propose the optical control of the biopolymer functions by a photoresponsive cationic comb-type copolymer. To prepare photoresponsive cationic comb-type copolymers, a photochromic molecule with ionic properties would be useful, because the ionic interaction of the copolymer with nucleic acids or peptides plays a pivotal role in chaperoning activity. Malachite green (MG) is converted from a colorless nonionic form to a green-colored cation form upon photoirradiation [Citation32]. The photoirradiation-dependent interactions of MG-bearing copolymers of acrylamide [Citation33,Citation34], N,N-dimethylacrylamide [Citation35], and vinyl alcohol [Citation36,Citation37] with DNA have been reported previously. Uda et al. demonstrated that the photoirradiated MG-derivatized copolymer of vinyl alcohol interacted with G-quadruplex DNA via electrostatic interactions and end stacking to increase the thermal stability of G-quadruplex DNA [Citation38].
In this study, a photoresponsive cationic comb-type copolymer was designed as a smart artificial chaperone for expanding applications of cationic comb copolymers. Previously, most of polymers with MG moiety was prepared by radical polymerization using a styrene derivative of MG as a monomer. Novel MG derivatives functionalized with carboxyl groups were synthesized to enable facile conjugation to the copolymer. To the best of our knowledge, the optical properties of MG derivative with a carboxyl group have not been investigated. Therefore, we first characterize photoresponsiveness of carboxyl derivatives of MG. The selected MG derivative was conjugated to a cationic comb-type copolymer. We examined the photoresponsivness of MG-modified cationic comb-type copolymers and demonstrated the photocontrol of membrane disruptive E5 peptide activity mediated by the copolymer.
2. Experimental section
2.1. General methods
Poly(allylamine) hydrochloride (PAA⋅HCl, Mw: 5 × 103) aqueous solution was kindly supplied by Nitto Boseki Co, Ltd. and was purified by precipitation in methanol. Poly(ethylene glycol) (PEG, Mn: 5 × 103) functionalized with aldehyde end group, 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) and N-(carbonylmethoxypolyethyleneglycol)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (PEG2KDSPE) were purchased from NOF Corporation. N-(Lissamine rhodamine B sulfonyl)-1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (LR-DOPE) was purchased from Avanti Polar Lipids. E5 was purchased from HiPep Laboratories. The sequence of E5 is H-GLFEAIAEFIEGGWEGLIEG-OH. Other reagents were purchased from Tokyo Chemical Industries, FUJIFILM Wako Pure Chemical Corporation, Watanabe Chemical Industries, and Nacalai Tesque. The course of reactions was monitored by thin-layer chromatography on silica gel plates (Silica Gel 60 F254). Wakogel 60 was used for silica gel column chromatography. NMR spectra were recorded on a Bruker AVANCE II 400 C (1H: 400 MHz, 13C: 100 MHz). ESI mass spectra were recorded with a Brucker microTOF II. UV-visible spectra were obtained at 200–800 nm using a JASCO V-730 UV/vis spectrophotometer. The concentration of E5 peptide was calculated using absorbance determined by UV-vis spectroscopy on a Shimadzu UV-1650PC using the molar extinction coefficient of tryptophan (5500 M−1 at 280 nm). Bis(4-(dimethylamino)phenyl)(4-ethynylphenyl)methanol (1) [Citation39], bis(4-(dimethylamino)phenyl)(phenyl)methanol (MG-OH) [Citation40], and bis(4-(dimethylamino)phenyl)(4-phenyl)methyl cyanide (MG-CN) [Citation32] were prepared according to the reported procedures. Photoirradiation was performed with a UVLED illuminator (wavelength: 310 ± 5 nm, LSP7NUV310-0024, from AITEC SYSTEM Co., Ltd.). The circular dichroism (CD) spectra were recorded on a JASCO J-820 CD spectrometer. Liposomes were studied using spinning disk confocal microscopy (CSU-X1, Yokogawa). The microscope (IX71, Olympus) was equipped with an EMCCD camera (iXon+ 893, Andor) and a 100× objective len (UPlanSApo NA 1.4, Olympus).
2.2. Synthesis of malachite green derivatives
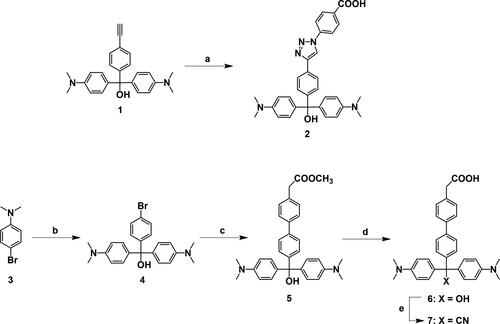
The malachite green derivatives including compounds 2, 6, and 7 were synthesized according to Scheme 1.
Scheme 1. Synthesis of MG derivatives functionalized with carboxylic acid. (a) 4-Azidobenzoic acid, DMF/water (2:1), room temperature, 65%; (b) n-BuLi, methyl 4-(bromomethyl)benzoate, THF, −65 °C, 92%; (c) 4-(methoxycarbonylmethyl)phenylboronic acid pinacol ester, Pd(PPh3)4, THF, reflux, 46%; (d) NaOH, DMSO/water (4:1), 40 °C, 87%; (e) HCl, KCN, DMSO/water (4:1), 60 °C, 37%.
2.2.1. Synthesis of compound 2 (MG 2)
The mixture of compound 1 (104 mg, 0.281 mmol), 4-azidobenzoic acid (53.1 mg, 0.321 mmol), L-ascorbic acid (10 mg, 0.0568 mmol), and copper sulfate (15.6 mg, 0.0624 mmol) in anhydrous N,N-dimethylformamide (2 ml) was stirred for 24 h. The mixture was extracted with chloroform, washed with water then brine, dried over anhydrous magnesium sulfate, and concentrated in vacuo. The crude product was purified by silica gel column chromatography (chloroform/methanol/acetic acid, 100:10:1) to give compound 2 (98 mg, 65%) as dark green oil. 1H NMR (400 MHz, DMSO-d6) δ 9.25 (s, 1H), 8.06 (d, J = 7.8 Hz, 2H), 7.90 (m, 4H), 7.46 (d, J = 8.4 Hz, 2H), 7.31 (d, J = 8.3 Hz, 2H), 7.14 (d, J = 8.8 Hz, 2H), 7.01 (d, J = 8.8 Hz, 2H), 6.65 (m, 4H), 5.97 (brs, 1H), 2.93 (s, 1H), 2.86 (d, J = 6.5 Hz, 12H). HRMS (ESI[+], CH3OH) m/z for C32H30N5O2 calcd. [M-OH]+: 516.2394; found: 516.2402.
2.2.2. Synthesis of compound 4
Compound 3 (3.01 g, 15.0 mmol) was dissolved in anhydrous tetrahydrofuran (THF) (40 ml) and the solution was kept at −65 °C in a methanol bath with dry ice. A hexane solution of n-butyllithium (11 mL, 17.3 mmol) was injected gradually into the THF solution, and the mixture was stirred for 30 min at −65 °C. Anhydrous THF (20 ml) solution of methyl-4-bromobenzoate (1.5 g, 6.98 mmol) was added dropwise to the mixture, and the mixture was allowed to warm slowly to room temperature and then stirred for 16 h. After the reaction was quenched by a saturated ammonium chloride aqueous solution, the mixture was extracted with ethyl acetate, washed with water then brine, dried over anhydrous magnesium sulfate, and concentrated in vacuo. The target compound was recrystallized from hexane three times, resulting in light green solid (2.77 g, 92%) with a melting point of 176–178 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.44 (d, J = 8.5 Hz, 2H), 7.13 (d, J = 8.5 Hz, 2H), 6.94 (d, J = 8.8 Hz, 4H), 6.61 (d, J = 8.9 Hz, 4H), 6.00 (brs, 1H), 2.84 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 149.6, 147.0, 134.9, 130.7, 129.6, 128.7, 120.7, 111.7, 81.2, 40.5. HRMS (ESI[-], CHCl3) m/z for C23H25BrClN2O calcd. [M + Cl]−: 461.0818; found: 461.0828.
2.2.3. Synthesis of compound 5
The mixture of compound 4 (502 mg, 1.18 mmol), tetrakis(triphenylphosphine)palladium (140 mg, 0.121 mmol), 4-(methoxycarbonylmethyl)phenylboronic acid pinacol ester (1.684 mmol), and potassium carbonate (670 mg, 4.85 mmol) in anhydrous THF (30 ml) was stirred at reflux for 24 h. The mixture was extracted with ethyl acetate, washed with water then brine, dried over anhydrous magnesium sulfate, and concentrated in vacuo. The crude product was purified by silica gel column chromatography (hexane/ethyl acetate, 1:2) to give compound 5 (266 mg, 46%) as green solid with a melting point of 68–70 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.59 (d, J = 8.2 Hz, 2H), 7.54 (d, J = 8.5 Hz, 2H), 7.32 (d, J = 8.3 Hz, 2H), 7.27 (d, J = 8.5 Hz, 2H), 7.00 (d, J = 8.9 Hz, 4H), 6.63 (d, J = 9.0 Hz, 4H), 5.93 (s, 1H), 3.70 (s, 2H), 3.61 (s, 3H), 2.85 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 172.1, 149.5, 147.1, 139.9, 138.9, 135.5, 132.8, 129.6, 128.8, 128.2, 127.3, 126.3, 111.7, 81.4, 52.1, 40.8, 40.6. HRMS (ESI, CH3CN) m/z for C32H33ClN2O3 calcd. [M − H]−: 493.2491; found: 493.2482.
2.2.4. Synthesis of compound 6 (MG 6)
A solution of compound 5 (70 mg, 0.142 mmol) in DMSO (4 ml) was added to an aqueous solution of NaOH (1 mL, 0.566 mmol) and was stirred at room temperature for 3 h. The mixture was extracted with chloroform, washed with water then brine, and concentrated in vacuo to give compound 6 (60 mg, 87%) as a green solid with a melting point of 154–156 °C. 1H NMR (400 MHz, DMSO-d6: D2O = 5: 1, (1% NaOD)) δ 7.50–7.45 (m, 4H), 7.28 (d, J = 8.2 Hz, 2H), 7.22 (d, J = 8.6 Hz, 2H), 6.98 (d, J = 8.9 Hz, 4H), 6.61 (d, J = 9.0 Hz, 4H), 3.14 (s, 2H), 2.82 (s, 12H); 13C NMR (400 MHz, DMSO-d6: D2O = 5: 1, (1% NaOD)) δ 175.2, 149.5, 148.2, 139.0, 138.6, 137.2, 136.5, 130.2, 128.9, 128.6, 126.3, 125.7, 111.9, 80.3, 49.0, 40.6. HRMS (ESI[-], CHCl3) m/z for C31H32ClN2O3 calcd. [M + Cl]−: 515.2101; found: 515.2100.
2.2.5. Synthesis of compound 7 (MG 7)
A solution of compound 6 (31 mg, 0.0645 mmol) in DMSO (4 ml) was added to 3 M HCl (250 µl) and then stirred for 30 min at 60 °C. To the mixture was added dropwise an aqueous solution of KCN (1 ml, 50 mg, 0.762 mmol), and the mixture was then stirred for 5 h at 60 °C. The resulting mixture was extracted with chloroform, washed with water then brine, dried over anhydrous magnesium sulfate, and concentrated under vacuum. The target compound was recrystallized from water three times, resulting in a green solid (13 mg, 37%) with a melting point of 228–229 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 7.69 (d, J = 8.5 Hz, 2H), 7.61 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.3 Hz, 2H), 7.21 (J = 8.5 Hz, 2H), 6.94 (d, J = 9.0 Hz, 4H), 6.73 (d, J = 9.0 Hz, 4H), 3.61 (s, 2H), 2.89 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 175.7, 149.9, 140.8, 139.9, 139.5, 132.6, 129.8, 129.5, 129.1, 128.2, 127.3, 127.0, 124.2, 112.1, 55.7, 40.5, 40.4. HRMS (ESI[-], CHCl3) m/z for C32H31ClN3O2 calcd. [M + Cl]−: 524.2105; found: 524.2098.
2.3. Preparation of cationic comb-type copolymers
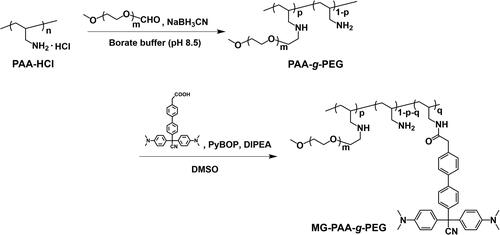
The cationic comb-type copolymers including PAA-g-PEG and MG-PAA-g-PEG were synthesized according to Scheme 2.
Scheme 2. Preparation of MG-modified cationic comb-type copolymers.
2.3.1. Preparation of PAA- g -PEG
The cationic comb-type copolymer poly(allylamine)-graft-poly(ethylene glycol) (PAA-g-PEG) was prepared as previously reported [Citation24]. Briefly, PAA-HCl and aldehyde-functionalized PEG were dissolved in 0.1 M borate buffer (pH 8.5). Sodium cyanoborohydride was added, and the solution was stirred for 4 days at 40 °C. The crude copolymer was purified by a cation exchange chromatography (TOYOPEARL SP-650) and was dialyzed against water using a cellulose membrane (MWCO: 3,500). The purified copolymer was obtained by freeze-drying. The obtained PAA-g-PEG consisted of 10 wt% PAA and 90 wt% PEG (9 mol% of allylamine units of PAA were substituted with PEG).
2.3.2. Preparation of MG-PAA-g-PEG
PAA-g-PEG modified with MG was prepared as follows: DIPEA (3 equiv. of the amino groups of PAA-g-PEG), various amounts of compound 7 and PyBOP (3 equiv. of compound 7 were added to PAA-g-PEG in DMSO and then stirred for 24 h at 40 °C. The crude copolymers were purified by dialysis against DMSO for 2 days and against water for 3 days using a cellulose membrane (MWCO: 3,500). The purified copolymers were obtained by freeze-drying. MG-modified PAA-g-PEG was characterized by 1H NMR.
2.4. Monitoring of photochemical reactions
Photochemical reactions of MG derivatives and MG-PAA-g-PEG were monitored on a JASCO V-730 UV/vis spectrophotometer. Absorption spectra of 10 µM MG derivatives either nonirradiated or photoirradiated at 310 nm (10 mW) were measured in 40 mM boric acid, 40 mM phosphoric acid, and 40 mM acetic acid (Britton-Robinson buffer) at pH 9.0 or pH 7.0 supplemented with 1% DMSO at 25 °C. The absorbance at 620 nm of MG derivatives before photoirradiation, Abs(0), was defined as 0% MG cationic form. MG derivatives were dissolved in Britton-Robinson buffer at pH 4.0 supplemented with 1% DMSO to obtain the maximum absorbance at 620 nm. Abs(∞), corresponds to 100% conversion to MG cationic form. The fraction of the conversion to MG cationic form was calculated from EquationEquation (1)(1)
(1) :
(1)
(1)
To calculate the fractions of MG derivative (compound 7) and MG modified with the cyano group (MG-CN), the absorbances at 620 nm of MG derivative (compound 6) and of unmodified MG (MG-OH), respectively, in Britton-Robinson buffer (pH 4.0) supplemented with 1% DMSO were regarded as Abs(∞). Absorption spectra of nonirradiated and photoirradiated MG-PAA-g-PEGs (10 µM (MG unit)) in Britton-Robinson buffer (pH 7.0) were measured at 25 °C. When the fraction of MG-PAA-g-PEGs was calculated, the absorbance at 630 nm of MG-PAA-g-PEG was used in Abs(t) and Abs(0), and the absorbance at 620 nm of MG 6 in Britton-Robinson buffer (pH 4.0) supplemented with 1% DMSO was regarded as Abs(∞).
2.5. Circular dichroism measurements
E5 (5 µM) was incubated with or without copolymer (50 µM in allylamine units) in 80 µL of 10 mM Tris-HCl (pH 7.4, 140 mM NaCl) at 37 °C for 5 min. The solution of MG-PAA-g-PEG was photoirradiated at 310 nm for 1 min and then added to the E5 solution. CD spectra were recorded on a JASCO J-820 CD spectrometer. Measurements were acquired using a quartz cell with a 1 mm path length. Each spectrum shown represents the average of 8 scans from 200 to 300 nm with a 0.1 nm resolution, obtained at 100 nm/min with a bandwidth of 1 nm at 25 °C. Helicity was calculated using EquationEquation (2)(2)
(2) as reported previously [Citation41]:
(2)
(2)
2.6. Preparation of giant unilamellar vesicles
Vesicles were prepared by the reversible-phase method reported by Tsumoto et al. [Citation42]. Briefly, to a solution of 1.2 mM DOPC in chloroform/diethyl ether (total 500 µL) containing 1.4 mol% PEG2K-DSPE and 0.2 mol% LR-DOPE was added an aqueous solution (500 µL) containing 10 mM HEPES (pH 7.4, 90 mM NaCl, 100 mM sucrose). After vigorous mixing, the resulting water/oil emulsion was centrifuged (20,000 × g) for 1 min at 20 °C. The upper phase (organic solvent) was removed, and the lower aqueous phase was retained. The resulting vesicles were washed at least three times with buffer A (10 mM HEPES buffer (pH 7.4, 140 mM NaCl)) and then suspended in buffer A. The concentration of the vesicles in suspension was determined using the LabAssay™ Phospholipid (Wako Ind.).
2.7. Observation of morphology of lipid membrane
For data shown in Figures 5 and S6, the E5 peptide (3 µL, 120 µM) was added to the vesicle suspension (6 µL, 1 mM), and then nonirradiated or photoirradiated MG-PAA-g-PEGs (3 µL, 0.6–3.6 mM (allylamine units)) was added to the mixture of vesicles and E5 peptide in buffer A. The mixture was incubated for 10 min at 37 °C and then dropped onto a coverslip. The morphology of lipid membranes was observed using a confocal laser scanning microscopy by excitation at 561 nm.
For data shown in Figures 6 and S7, the vesicle suspension (12 µL, 1 mM) was incubated with E5 peptide (6 µL, 120 µM) in buffer A for 5 min at 37 °C. The mixture of vesicles and E5 was incubated with nonirradiated MG-PAA-g-PEGs (6 µL, 0.6–3.6 mM (allylamine units)) in buffer A for 5 min. The solution was divided in half. One sample was photoirradiated at 310 nm for 1 min and incubated in buffer A for 5 min. The other was incubated in buffer A for 6 min without photoirradiation. Morphologies of lipid membranes were immediately observed using confocal laser scanning microscopy by excitation at 561 nm.
2.8. Dissociation of photoirradiated MG-derivatized copolymer from the complex by the addition of poly(vinylsulfonate)
E5 peptide (3 µL, 120 µM) was added to the vesicle suspension (6 µL, 1 mM), and then nonirradiated or photoirradiated MG-PAA-g-PEG (1.5 µL, 2.4 mM, allylamine units) was added to the mixture of vesicles and E5 peptide in buffer A. After the incubation for 10 min at 37 °C, poly(vinylsulfonate) (PVS; 1.5 µL, 4.8 mM, sulfonate groups) was added to the solution to dissociate the complex of MG-derivatized copolymer and E5 peptide.
3. Results and discussion
3.1. Synthesis and photoionization of MG derivatives with carboxylic acid
We prepared two MG derivatives functionalized with carboxylic acid, compounds MG 2 and MG 6 (Scheme 1). MG 2 was synthesized by a click reaction between 4-azidobenzoic acid and compound 1, which was prepared according to a previously reported procedure [Citation39]. MG 6 was synthesized in three steps from compound 3: Compound 4 was formed from compound 3 by a Grignard reaction with methyl 4-bromobenzoate. Compound 5 was obtained by Suzuki-Miyaura coupling with 4-(methoxycarbonylmethyl)phenylboronic acid pinacol ester. The ester group of compound 5 was hydrolyzed under basic conditions to produce MG 6.
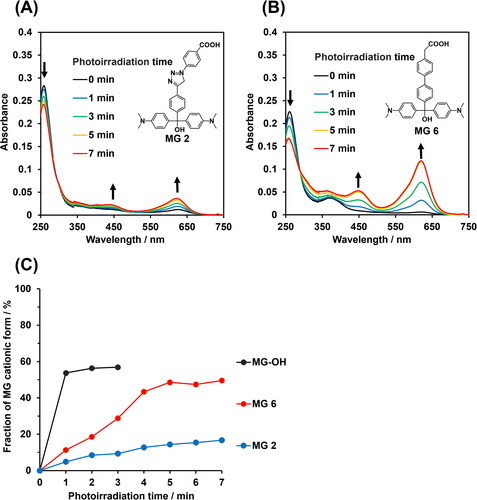
We investigated photoresponsive properties of synthesized MG 2 and MG 6 in aqueous solution at pH 9.0 using UV-vis spectroscopy. Before photoirradiation, the absorption peak of the MG derivatives was observed at around 260 nm, which was assigned to the nonionic form (). Photoirradiation at 310 nm resulted in decreases of absorbance at 260 nm and increases of absorbance at 450 and 620 nm (), indicating that the MG derivatives were converted from the nonionic to the cationic forms upon photoirradiation. The spectral changes are similar to that of unmodified MG molecule (MG-OH, Figure S1(A)). The fraction of the cationic form increased with increasing irradiation time and plateaued at 59, 19, and 49% for unmodified MG-OH, MG 2, and MG 6, respectively (). The low photoconversion of MG 2 compared to MG-OH and MG 6 is likely due to the extensive π-electron conjugation over the MG and the phenyl triazole units of MG 2.
Figure 1. Photoresponsive properties of MG derivatives. (A,B) Absorption spectral changes of 10 µM (A) MG 2 and (B) MG 6 in Britton-Robinson buffer (pH 9.0) supplemented with 1% DMSO upon photoirradiation at 310 nm. Arrows indicate direction of absorbance change as a function of irradiation time. (C) Fractions of MG cationic as a function of photoirradiation time.
It is known that MG derivatives with either hydroxyl or methoxy as a leaving group undergo cationization as pH decreases (Figure S2(A)) [Citation43,Citation44]. The increase of the absorbance at 620 nm as a function of the time of incubation of MG 6 at pH 7.0 without irradiation indicated that this compound was gradually converted to the cationic form (Figure S2(B)). This property of MG 6 means that it cannot be photoresponsively controlled under physiological conditions.
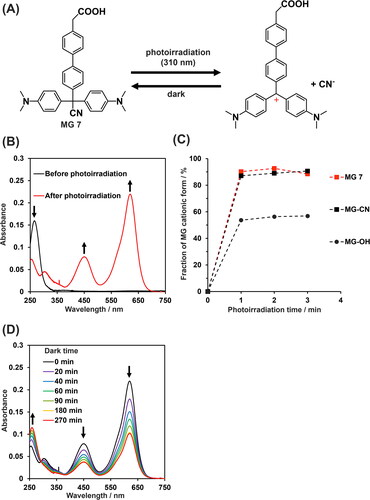
Next, we synthesized MG 7, which has cyano as a leaving group (Scheme 1). MG 7 did not spontaneously form a cation at pH 7.0 (Figure S2(C)), consistent with a previous report that MG functionalized with a cyano group is pH insensitive [Citation44]. MG 7 was rapidly converted to the cationic form by photoirradiation (). After 1 min of irradiation, we observed 92% conversion to the cationic form, which is similar to the percent conversion observed for MG molecule with cyano group after 1 min (). The reversibility of ionization of MG 7 was assessed since MG derivatives are known to revert from cationic forms to nonionic forms in the dark [Citation45]. The absorbance at 620 nm of cationic MG 7 decreased over time in the dark, indicative of reversion to the nonionic form (). Subsequent re-irradiation converted MG 7 to the cationic form, but the conversion was lower than that of the virgin sample (Figure S3).
Figure 2. Photoresponsive properties of MG 7. (A) Illustration of the reversible photochemical reaction of MG 7. (B) Absorbance spectra of 10 µM MG 7 before and after photoirradiation at 310 nm for 2 min. (C) Fraction of MG derivatives in cationic form as a function of photoirradiation time. (D) Absorbance spectra of 10 µM cationic MG 7 in the dark at 25 °C as a function of time (from top to bottom: 0, 20, 40, 60, 90, 180, and 270 min). buffer for all experiments was Britton-Robinson buffer (pH 7.0) supplemented with 1% DMSO.
3.2. Preparation and photoionization of MG-modified cationic comb-type copolymer
Encouraged by the high photoresponsiveness of MG 7, we synthesized the MG-modified cationic comb-type copolymers with the goal of using photoionization of MG 7 to control the chaperone activity of the copolymer (Scheme 2). The cationic comb-type copolymer PAA-g-PEG was prepared according to a reported procedure [Citation24]. MG 7 was conjugated to PAA-g-PEG by condensation between the amino groups of PAA-g-PEG and the carboxyl group of MG 7 using PyBOP as a condensation reagent. Three PAA-g-PEG/MG conjugates (MG-PAA-g-PEGs) that differed in MG content were prepared: 9 mol% MG (9MG9P), 21 mol% MG (21MG9P), and 41 mol% MG (41MG9P). We regulated the MG modification degree of MG-PAA-g-PEGs <50 mol% because high MG modification was expected to cause aggregation of MG groups and reduce photo responsiveness of the copolymer. Samples are referred to as XMGYP, where X and Y represent the mol% of the MG unit and the PEG unit, respectively (). The recovery and coupling efficiency of MG-PAA-g-PEGs were more than 85 and 80%, respectively.
Table 1. Compositions and properties of cationic comb-type copolymers.
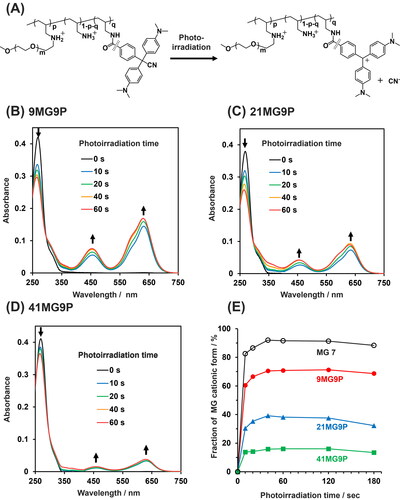
We next evaluated the photoconversions of MG-PAA-g-PEGs (). Before photoirradiation, MG-PAA-g-PEGs had absorption peaks at 269 nm derived from the nonionic form of the MG moiety. After photoirradiation, the height of this peak decreased, and the peak derived from the cationic form of the MG moiety appeared at 630 nm (). This result suggests that the MG moiety of MG-PAA-g-PEGs was converted from the nonionic to the cationic form by photoirradiation. Compared with MG 7, the absorption peaks observed for MG-PAA-g-PEGs showed a red shift (Figure S4), suggesting that there is π–π stacking between MG moieties in the copolymers. The conversion of MG-PAA-g-PEGs reached equilibrium after 40 s of photoirradiation. The maximum photoconversions to cationic forms of 9MG9P, 21MG9P, and 41MG9P were 71, 39, and 16%, respectively (). Thus, the extent of photoconversion decreased with an increase in MG content. We speculate that interactions between MG moieties in the copolymer could prevent photo-induced cationization.
Figure 3. The photoresponsive properties of MG-PAA-g-PEG. (A) Illustration of the photo-mediated conversion of MG-PAA-g-PEG to the cation form. (B–D) Absorbance spectra of 10 µM MG-PAA-g-PEGs (in MG units) (B) 9MG9P, (C) 21MG9P, and (D) 41MG9P as a function of time of photoirradiation at 310 nm. Buffer for all experiments was Britton-Robinson buffer (pH 7.0). (E) Fraction of MG derivatives in cationic form as a function of photoirradiation time.
3.3. Photoresponsive chaperone activity of MG-PAA-g-PEG toward E5 peptide
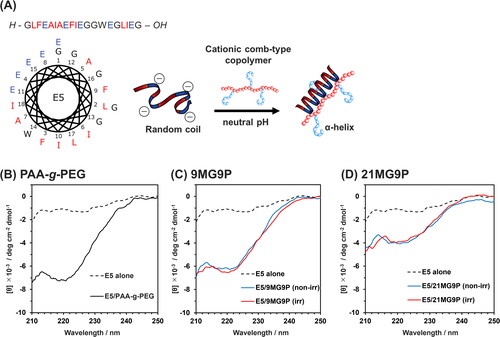
The effect of MG-PAA-g-PEG on the conformation of E5 peptide was investigated using CD spectroscopy. At pH 7.4, the E5 peptide is unstructured, but in the presence of PAA-g-PEG, E5 adopts a helical structure (), as previously reported [Citation22]. In the presence of PAA-g-PEG or nonirradiated MG-PAA-g-PEG, a negative CD signal due to E5 was observed in the wavelength region from 210 to 230 nm (); this band was assigned to the α-helix structure. The helicities of the peptide with PAA-g-PEG, nonirradiated 9MG9P, or nonirradiated 21MG9P were 15, 13, and 5%, respectively. Thus, MG modification of the copolymer reduced the helicity of E5, which indicates that E5 folding is affected by hydrophobic interaction between MG groups of the copolymers and hydrophobic residues of E5. However, E5 undergoes partial folding through hydrophobic interactions with lipid membranes [Citation23,Citation46]. Therefore, it isn’t not necessary that E5 folding is inhibited by the hydrophobic interaction. No obvious differences were observed between the CD spectra of E5 with nonirradiated MG-PAA-g-PEG and with photoirradiated MG-PAA-g-PEG (). These data indicate that the cationization of MG units did not contribute to E5 folding.
Figure 4. Effect of MG-PAA-g-PEG on the conformation of E5 peptide. (A) The conformation of E5 in a helical wheel diagram and illustration of conformation change induced by the cationic comb-type copolymer. (B–D) CD spectra of 5 µM E5 peptide incubated without or with nonirradiated or photoirradiated (B) PAA-g-PEG, (C) 9MG9P, and (D) 21MG9P (50 µM, allylamine units) for 10 min at 37 °C. Buffer for all experiments was 10 mM Tris-HCl buffer (pH 7.4, 140 mM NaCl).
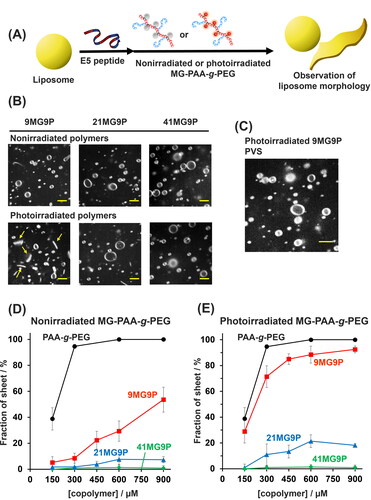
Next, we examined the effect of MG-PAA-g-PEG on the membrane-disruptive activity of E5 peptide using giant unilamellar vesicles (GUVs) as a model membrane (). When nonirradiated MG-PAA-g-PEG (300 µM, allylamine units) was added to a GUV suspension with 30 µM E5, vesicle structures were not altered compared to the sample lacking MG-PAA-g-PEG ( and Figure S5). Interestingly, when 300 µM (in allylamine units) photoirradiated MG-PAA-g-PEG was added to the GUVs and E5, the vesicle morphology changed in a manner that depended on the MG content of the copolymer: Photoirradiated 9MG9P induced a vesicle-to-sheet transformation, whereas photoirradiated 21MG9P and 41MG9P did not (). The addition of anionic PVS, which dissociates the copolymer from E5 [Citation24], caused a transition from lipid sheets into vesicles (). Therefore, the formation of an interpolyelectrolyte complex of 9MG9P and E5 was necessary for the vesicle-to-sheet transformation. Analysis of the concentration dependence of MG-PAA-g-PEG derivatives on the vesicle-to-sheet transformation confirmed that the transformation activity of the copolymers was reduced with an increase in MG content ( and Figure S6). The photoirradiation of 9MG9P resulted in sheet formation (), indicating that the chaperone activity of the copolymer can be photocontrolled. E5 is localized at the surface of liposome via hydrophobic interactions and is folded and clustered by cationic comb-type copolymers. Copolymer/E5 complexes assemble at the exposed membrane edge to stabilize the interface between the edge and solution, leading to lipid sheet formation. We previously reported that an increase in hydrophobicity of the copolymers decreases the localization of the complexes to edges of lipid sheets [Citation47]. We consider that photo-induced cationization of 9MG9P induced the enhancement of the hydrophilicity of 9MG9P/E5 complex and stabilization of edges of lipid membrane for the photoconversion of liposome morphology.
Figure 5. Analysis of effect of MG-derivatized copolymer on E5 activity. (A) Schematic of the morphological conversion of lipid membranes induced by MG-PAA-g-PEG. (B) Confocal microscopic images of GUVs treated with 30 µM E5 peptide and 300 µM nonirradiated or photoirradiated MG-PAA-g-PEG (in allylamine units) at 37 °C. (C) Image of vesicles treated with 30 µM E5 and 300 µM 9MG9P (in allylamine units) at 37 °C and then 600 µM PVS (in anion units) for 1 min. Scale bars: 5 µm. (D,E) The percentage of lipid sheets when GUVs were treated with 30 µM E5 over a range of concentrations of (D) nonirradiated or (E) photoirradiated MG-PAA-g-PEG for 10 min at 37 °C. Buffer for all experiments was 10 mM HEPES (pH 7.4), 140 mM NaCl.
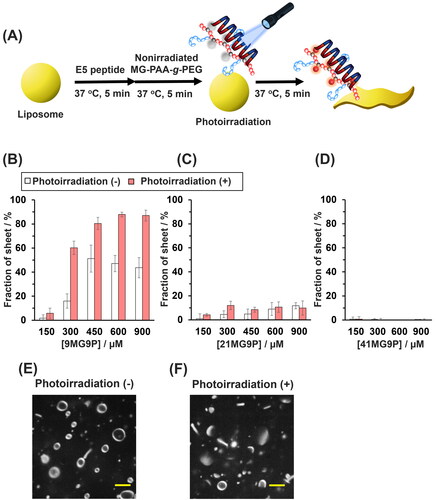
Finally, in situ photoregulation of E5 activity by MG-PAA-g-PEG was explored. GUVs were mixed with nonirradiated MG-PAA-g-PEG and E5 and were photoirradiated for 1 min (). A significant increase in the percent of lipid sheets was observed in the presence of 300 to 900 µM 9MG9P (in allylamine units), whereas no morphological changes in lipid membranes were observed in the presence of 21MG9P or 41MG9P (, Figure S7). At 300 µM 9MG9P (in allylamine units), a 40% increase in the percentage of lipids sheets was triggered by the photoirradiation (). No morphological changes were observed when 9MG9P alone or E5 alone was added to GUVs with or without photoirradiation (Figure S8). Hence, in situ photoresponsive activation of E5 peptide was achieved with 9MG9P.
Figure 6. In situ photoresponsive activation of E5 peptide by 9MG9P. (A) Schematic of the morphological photoconversion of lipid membranes induced by 9MG9P/E5. (B–D) The percentage of lipid sheets when GUVs were treated with 30 µM E5 peptide and indicated concentrations of (B) 9MG9P, (C) 21MG9P, and (D) 41MG9P were nonirradiated or photoirradiated at 310 nm for 1 min. (E,F) Confocal microscopic images of GUVs mixed with 30 µM E5 and 300 µM 9MG9P (in allylamine units) and (E) photoirradiated at 310 nm or (F) not for 1 min. Scale bars: 5 µm. Buffer for all experiments was 10 mM HEPES (pH 7.4), 140 mM NaCl.
4. Conclusion
In conclusion, we prepared a novel photoresponsive cationic comb-type copolymer as a smart artificial chaperone. Two types of MG carboxylic acid derivatives that could be easily conjugated with polymers were synthesized. The π-electron conjugation structure of the MG influenced photo-conversion of MG derivatives. MG 7 was quickly and quantitatively transformed to cationic form by photoirradiation. MG 7 was conjugated to PAA-g-PEG using a condensation reaction. UV spectrometry revealed that the photoresponsiveness of MG in the conjugated form was reduced as MG content was increased. MG-PAA-g-PEG bearing 9 mol% MG enhanced the membrane disruptive activity of E5 peptide and induced morphological changes in liposomes in a manner that could be controlled by photoirradiation. The mechanism underlying the photo-mediated enhancement of E5 activity by 9MG9P remains to be determined. 9MG9P promoted a change in E5 to a helical form regardless of photoirradiation as assessed by CD spectroscopy. We speculate that the formation of the MG cation in the MG-PAA-g-PEG conjugate affects the assembly and localization of E5 peptides on lipid membranes since the multimolecular organization of E5 peptides at lipid sheet edges is necessary for the stabilization of lipid sheets [Citation24,Citation47]. Despite causing structural disruption of biomacromolecules, UV light has been employed to achieve photoregulation of biomacromolecular functions through the introduction of responsive molecules such as azobenzene [Citation48,Citation49], diarylethene [Citation50,Citation51] and caged compounds [Citation52,Citation53]. Therefore, we assume that the use of UV light does not affect the functional control of E5. The MG derivative with a carboxy group could be used to impart photoresponsiveness to various materials such as biopolymers and inorganic materials.
Supplemental Material
Download MS Word (3.5 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Radford SE, Dobson CM. From computer simulations to human disease: emerging themes in protein folding. Cell. 1999;97(3):291–298. doi: 10.1016/s0092-8674(00)80739-4.
- Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81(11):678–699. doi: 10.1007/s00109-003-0464-5.
- Knowles T, Vendruscolo M, Dobson C. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15(6):384–396. doi: 10.1038/nrm3810.
- Mayhew M, da Silva ACR, Martin J, et al. Protein folding in the central cavity of the GroEL–GroES chaperonin complex. Nature. 1996;379(6564):420–426. doi: 10.1038/379420a0.
- Hartl MH, Bracher A, Hartl FU. The GroEL-GroES chaperonin machine: a nano-cage for protein folding. Trends Biochem Sci. 2016;41(1):62–76. doi: 10.1016/j.tibs.2015.07.009.
- Akiyoshi K, Sasaki Y, Sunamoto J. Molecular chaperone-like activity of hydrogel nanoparticles of hydrophobized pullulan: thermal stabilization with refolding of carbonic anhydrase B. Bioconjug Chem. 1999;10(3):321–324. doi: 10.1021/bc9801272.
- Beierle JM, Yoshimatsu K, Chou B, et al. Polymer nanoparticle hydrogels with autonomous affinity switching for the protection of proteins from thermal stress. Angew Chem Int Ed Engl. 2014;53(35):9275–9279. doi: 10.1002/anie.201404881.
- Liu X, Liu Y, Zhang Z, et al. Temperature-responsive mixed-shell polymeric micelles for the refolding of thermally denatured proteins. Chemistry. 2013;19(23):7437–7442. doi: 10.1002/chem.201300634.
- Maruyama A, Katoh M, Ishihara T, et al. Comb-type polycations effectively stabilize DNA triplex. Bioconjug Chem. 1997;8(1):3–6. doi: 10.1021/bc960071g.
- Maruyama A, Watanabe H, Ferdous A, et al. Characterization of interpolyelectrolyte complexes between double-stranded DNA and polylysine comb-type copolymers having hydrophilic side chains. Bioconjug Chem. 1998;9(2):292–299. doi: 10.1021/bc9701510.
- Kim WJ, Sato Y, Akaike T, et al. Cationic comb-type copolymers for DNA analysis. Nat Mater. 2003;2(12):815–820. doi: 10.1038/nmat1021.
- Kim WJ, Sato Y, Akaike T, et al. DNA strand exchange stimulated by spontaneous complex formation with cationic comb-type copolymer. J Am Chem Soc. 2002;124(43):12676–12677. doi: 10.1021/ja0272080.
- Shimada N, Saito K, Miyata T, et al. DNA computing boosted by a cationic copolymer. Adv Funct Mater. 2018;28:1707406.
- Makita N, Inoue S, Akaike T, et al. Improved performance of a DNA nanomachine by cationic copolymers. Nucleic Acids Symp Ser. 2004;48(48):173–174. doi: 10.1093/nass/48.1.173.
- Wang J, Shimada N, Maruyama A. Cationic copolymer-augmented DNA hybridization chain reaction. ACS Appl Mater Interfaces. 2022;14(34):39396–39403. doi: 10.1021/acsami.2c11548.
- Gao J, Shimada N, Maruyama A. Enhancement of deoxyribozyme activity by cationic copolymers. Biomater Sci. 2015;3(2):308–316. doi: 10.1039/c4bm00256c.
- Hanpanich O, Oyanagi T, Shimada N, et al. Cationic copolymer-chaperoned DNAzyme sensor for microRNA detection. Biomaterials. 2019;225:119535. doi: 10.1016/j.biomaterials.2019.119535.
- Rudeejaroonrung K, Hanpanich O, Saito K, et al. Cationic copolymer enhances 8–17 DNAzyme and MNAzyme activities. Biomater Sci. 2020;8(14):3812–3818. doi: 10.1039/d0bm00428f.
- Hanpanich O, Saito K, Shimada N, et al. One-step isothermal RNA detection with LNA-modified MNAzymes chaperoned by cationic copolymer. Biosens Bioelectron. 2020;165:112383. doi: 10.1016/j.bios.2020.112383.
- Takahashi S. Conformation of membrane fusion-active 20-residue peptides with or without lipid bilayers. Implication of alpha-helix formation for membrane fusion. Biochemistry. 1990;29(26):6257–6264. doi: 10.1021/bi00478a021.
- Murata M, Takahashi S, Shirai Y, et al. Specificity of amphiphilic anionic peptides for fusion of phospholipid vesicles. Biophys J. 1993;64(3):724–734. doi: 10.1016/S0006-3495(93)81432-2.
- Shimada N, Kinoshita H, Tokunaga S, et al. Inter-polyelectrolyte nano-assembly induces folding and activation of functional peptides. J Control Release. 2015;218:45–52. doi: 10.1016/j.jconrel.2015.10.001.
- Sakamoto W, Ochiai T, Shimada N, et al. Cationic copolymer augments membrane permeabilizing activity of an amphiphilic peptide. J Biomater Sci Polym Ed. 2017;28(10–12):1097–1108. doi: 10.1080/09205063.2017.1293483.
- Shimada N, Kinoshita H, Umegae T, et al. Cationic copolymer-chaperoned 2D–3D reversible conversion of lipid membranes. Adv Mater. 2019;31(44):e1904032. doi: 10.1002/adma.201904032.
- Bertelson RC. In: Brown GH, editor. Techniques of chemistry, III. New York (NY): Wiley-Interscience; 1971.
- Kotharangannagari VK, Sànchez-Ferrer A, Ruokolainen J, et al. Photoresponsive reversible aggregation and dissolution of rod-coil polypeptide diblock copolymers. Macromolecules. 2011;44(12):4569–4573. doi: 10.1021/ma2008145.
- Rahimi S, Stumpf S, Grimm O, et al. Dual photo- and pH-responsive spirooxazine-functionalized dextran nanoparticles. Biomacromolecules. 2020;21(9):3620–3630. doi: 10.1021/acs.biomac.0c00642.
- Higuchi A, Hamamura A, Shindo Y, et al. Photon-modulated changes of cell attachments on poly(spiropyran-co-methyl methacrylate) membranes. Biomacromolecules. 2004;5(5):1770–1774. doi: 10.1021/bm049737x.
- He D, Arisaka Y, Masuda K, et al. A photoresponsive soft interface reversibly controls wettability and cell adhesion by conformational changes in a spiropyran-conjugated amphiphilic block copolymer. Acta Biomater. 2017;51:101–111. doi: 10.1016/j.actbio.2017.01.049.
- Xiao X, Hu J, Wang X, et al. A dual-functional supramolecular hydrogel based on a spiropyran–galactose conjugate for target-mediated and light-controlled delivery of microRNA into cells. Chem Commun. 2016;52(84):12517–12520. doi: 10.1039/c6cc07386g.
- Hirakura T, Nomura Y, Aoyama Y, et al. Photoresponsive nanogels formed by the self-assembly of spiropyrane-bearing pullulan that act as artificial molecular chaperones. Biomacromolecules. 2004;5(5):1804–1809. doi: 10.1021/bm049860o.
- Herz ML. Photochemical ionization of the triarylmethane leuconitriles. J Am Chem Soc. 1975;97(23):6777–6785. doi: 10.1021/ja00856a029.
- Irie M, Kunwatchakun D. Photoresponsive polymers. 8. Reversible photostimulated dilation of polyacrylamide gels having triphenylmethane leuco derivatives. Macromolecules. 1986;19(10):2476–2480. doi: 10.1021/ma00164a003.
- Kito H, Suzuki F, Nagahara S, et al. A total delivery system of genetically engineered drugs or cells for diseased vessels. Asaio J. 1994;40(3):M260–M266. doi: 10.1097/00002480-199407000-00005.
- Umeda M, Harada-Shiba M, Uchida K, et al. Photo-control of the polyplexes formation between DNA and photo-cation generatable water-soluble polymers. Curr Drug Deliv. 2005;2(3):207–214. doi: 10.2174/1567201054367986.
- Uda RM, Ohshita M. Phototriggered DNA complexation and compaction using poly(vinyl alcohol) carrying a malachite green moiety. Bio Macromolecules. 2012;13(5):1510–1514. doi: 10.1021/bm3001952.
- Uda RM, Matsui T. Photoinduced conformational changes in DNA by poly(vinyl alcohol) carrying a malachite green moiety for protecting DNA against attack by nuclease. Soft Matter. 2015;11(42):8246–8252. doi: 10.1039/c5sm01874a.
- Uda RM, Nishimoto N, Matsui T, et al. Photoinduced binding of malachite green copolymer to parallel G-quadruplex DNA. Soft Matter. 2019;15(22):4454–4459. doi: 10.1039/c9sm00411d.
- Oßwald S, Breimaier S, Linseis M, et al. Polyelectrochromic vinyl ruthenium-modified tritylium dyes. Organometallics. 2017;36(10):1993–2003. doi: 10.1021/acs.organomet.7b00194.
- Tachikawa T, Handa C, Tokita S. Synthesis and radiation sensitivity of tris(4-N,N-dimethylainophenyl)methanethiol. J Photopol Sci Technol. 2003;16(2):187–190. doi: 10.2494/photopolymer.16.187.
- Sakamoto W, Masuda T, Ochiai T, et al. Cationic copolymers act as chaperones of a membrane-active peptide: influence on membrane selectivity. ACS Biomater Sci Eng. 2019;5(11):5744–5751. doi: 10.1021/acsbiomaterials.8b01582.
- Tsumoto K, Hayashi Y, Tabata J, et al. A reverse-phase method revisited: rapid high-yield preparation of giant unilamellar vesicles (GUVs) using emulsification followed by centrifugation. Colloid Surf A. 2018;546:74–82. doi: 10.1016/j.colsurfa.2018.02.060.
- Culp SJ, Beland FA. Malachite green: a toxicological review. J Am Coll Toxicol. 1996;15(3):219–238. doi: 10.3109/10915819609008715.
- Uda RM, Yoshida N, Iwasaki T, et al. pH-triggered solubility and cytotoxicity changes of malachite green derivatives incorporated in liposomes for killing cancer cells. J Mater Chem B. 2020;8(36):8242–8248. doi: 10.1039/d0tb01346c.
- Jiang Y, Wan P, Xu H, et al. Facile reversible UV-controlled and fast transition from emulsion to gel by using a photoresponsive polymer with a malachite green group. Langmuir. 2009;25(17):10134–10138. doi: 10.1021/la900916m.
- Hsu CH, Wu SH, Chang DK, et al. Structural characterizations of fusion − peptide analogs of influenza virus hemagglutinin: implication of the necessity of a helix-hinge-helix motif in fusion activity. J Biol Chem. 2002;277(25):22725–22733. doi: 10.1074/jbc.M200089200.
- Masuda T, Takahashi S, Ochiai T, et al. Autonomous vesicle/sheet transformation of cell-sized lipid bilayers by hetero-grafted copolymers. ACS Appl Mater Interfaces. 2022;14(48):53558–53566. doi: 10.1021/acsami.2c17435.
- Asanuma H, Liang X, Nishioka H, et al. Synthesis of azobenzene-tethered DNA for reversible photo-regulation of DNA functions: hybridization and transcription. Nat Protoc. 2007;2(1):203–212. doi: 10.1038/nprot.2006.465.
- Goldau T, Murayama K, Brieke C, et al. Azobenzene C-Nucleosides for photocontrolled hybridization of DNA at room temperature. Chemistry. 2015;21(49):17870–17876. doi: 10.1002/chem.201503303.
- Babii O, Afonin S, Berditsch M, et al. Controlling biological activity with light: diarylethene-containing cyclic peptidomimetics. Angew Chem. 2014;126(13):3460–3463. doi: 10.1002/ange.201310019.
- Kneuttinger AC, Winter M, Simeth NA, et al. Artificial light regulation of an allosteric bienzyme complex by a photosensitive ligand. Chembiochem. 2018;19(16):1750–1757. doi: 10.1002/cbic.201800219.
- Ando H, Furuta T, Tsien RY, et al. Photo-mediated gene activation using caged RNA/DNA in zebrafish embryos. Nat Genet. 2001;28(4):317–325. doi: 10.1038/ng583.
- Karginov AV, Zou Y, Shirvanyants D, et al. Light regulation of protein dimerization and kinase activity in living cells using photocaged rapamycin and engineered FKBP. J Am Chem Soc. 2011;133(3):420–423. doi: 10.1021/ja109630v.