
Abstract
Objective
This study aimed to identify novel pathogenic genes and variants in a Chinese family with premature ovarian insufficiency (POI).
Methods
A Chinese POI family was enrolled in this study. Whole exome sequencing was performed on the proband and her mother to identify the potential causative genes and variants and Sanger sequencing was used to confirm the finally identified potential pathogenic variant in the family.
Results
An assessment of the family pedigree suggested that POI was inherited in an autosomal dominant manner in this family. A novel missense variant of the laminin subunit gamma-1 gene (LAMC1; NM_002293.4: c.3281A > T, p.D1094V) was finally identified in the proband and her affected mother. This variant was not found in any public databases. In silico analysis indicated the amino acid encoded at the variant site was highly conserved among mammals and associated with decreased protein stability and disrupted protein function. Its presence in the POI family was confirmed by Sanger sequencing.
Conclusions
This study firstly reported a novel missense variant of LAMC1 in a Chinese POI family, which was inherited in an autosomal dominant manner. This variant may result in the development of POI. Our results provide supporting evidence for a causative role for LAMC1 variants in POI.
Introduction
Premature ovarian insufficiency (POI) is a serious reproductive disorder characterized by the depletion or loss of normal ovarian function in women under 40 years of age. It affects about 1% of women of childbearing age worldwide, and typically manifests with a ≥ 4-month history of oligomenorrhea/amenorrhea and follicle-stimulating hormone (FSH) levels >25 IU/L in two measurements for at least 4 weeks [Citation1,Citation2]. Hormone replacement therapy (HRT) can partially alleviate the symptoms caused by POI, but there are limited effective treatments for the reproductive impairment [Citation3].
The etiologies of POI are complex, and mainly including genetic, environmental, autoimmune, or other factors [Citation4]; however, the causes of most POI cases remain unclear. Several studies have suggested that genetic factors play an important role in POI, and various genes have been identified to harbor variants that affect the biological function associated with POI [Citation5]. With the rapid development of sequencing technology, whole-exome sequencing (WES) is now widely used, and has proven useful in POI gene discovery [Citation6]. Multiple genes associated with POI have been identified in affected families using WES, including BRCA2, PSMC3IP, SYCP2L, BUB1B, STAG3, SPIDR, POLR2C, MEIOB, MCM8, MCM9, MRPS22, FIGLA, and PMM2 [Citation7–17]. Similarly, WES has identified genes associated with POI in sporadic patients, including NANOS3, BNC1, EIF2B4, FOXL2, FANCA, SALL4, EIF2B3, and GHR [Citation6,Citation18–21].
Considering that POI is a highly heterogeneous disease, a better understanding of the underlying mechanisms and genetic etiology is needed. Additionally, very few families with autosomal dominant POI have been described and assessed by WES to identify causative genes. Here, we performed WES in a Chinese family with POI and identified a novel rare missense variant (c.3281A > T, p.D1094V) of the laminin subunit gamma-1 gene (LAMC1; NM_002293.4). Our findings expand the spectrum of POI causative variants, which may be useful for future pathogenic studies.
Materials and methods
Study subjects
A Chinese family with POI was enrolled in this study from the Acupuncture and Moxibustion Hospital of the China Academy of Chinese Medical Sciences. The proband was diagnosed with POI according to previously described criteria [Citation22]: oligomenorrhea/amenorrhea for at least 4 months, and an elevated FSH level >25 IU/L on two occasions >4 weeks apart. Her mother started irregular menstrual cycles at 34 years of age and developed amenorrhea at the age of 40 years. Both patients have a normal 46, XX karyotype. Five ml of peripheral blood was collected from the proband and her parents. This study was in accordance with the principles of the Declaration of Helsinki, and was approved by the ethics committee of the Institute of Acupuncture and Moxibustion, China Academy of Chinese Medical Sciences. Written informed consent was obtained from all participants.
DNA extraction
Genomic DNA was extracted from peripheral blood using a QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The quality of the DNA samples was assessed using a NanoDrop2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA).
WES analysis
WES was performed on the proband and her mother. The patients’ exomes were captured using a SureSelect Human All Exon V6 Enrichment kit (Agilent, Santa Clara, CA), and then sequenced on a NovaSeq platform (Illumina, San Diego, CA) according to the manufacturer’s guidance. All reads were mapped to the human reference genome (hg19) using Burrows–Wheeler Alignment version 0.7.9a (http://bio-bwa.sourceforge.net). Single nucleotide variants and indels were detected using Genome Analysis Toolkit version 3.5 software (https://gatk.broadinstitute.org/hc/en-us) and annotated using ANNOVAR (https://annovar.openbioinformatics.org/en/latest/user-guide/download/) by consideration of splice site, intronic, exonic, 5′ untranslated region (UTR), 3′ UTR, intergenic, upstream, or downstream locations.
Variant filtering
Heterozygous variants shared by the proband and her affected mother were selected according to the following criteria: i) missense, nonsense, frameshift, non-frameshift, or splicing site variants; ii) variants with a MAF <0.1% in East Asians and the total population in the 1000 Genomes Project (1KG Project; http://browser.1000genomes.org) and GnomAD (https://gnomad.broadinstitute.org) dataset. Predictions of deleterious nonsynonymous variants were performed using four following online software programs: SIFT (http://sift-dna.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), MutationTaster (http://www.mutationtaster.org), and CADD (http://cadd.gs.washington.edu), which integrates information from various functional annotations and presents this as a score. CADD pathogenicity prediction scoring >20 predicts the top 1% of deleterious variants [Citation23]. The final candidate variant was evaluated according to ACMG variant interpretation guidelines.
Sanger sequencing validation
The final candidate variant was validated in family members using standard Sanger sequencing. Primer5 software was used to design specific primers which are shown in .
Alignment of the LAMC1 protein
Alignment of the LAMC1 protein among different species (human, mouse, rat, cattle, wild pig, Xenopus, and medaka) was performed using CLC Sequence Viewer 8 software to investigate sequence conservation.
Protein structure stability prediction
To determine the effect of the variant on protein structure and function, we used two different protein stability prediction tools namely I-Mutant2.0 [Citation24], Impact of Non synonymous variations on Protein Stability-Multi-Dimension (INPS-MD) [Citation25]. The protein sequence of LAMC1 was retrieved from the NCBI protein database and was used for the analysis of protein stability. The accession numbers for the protein sequence of LAMC1 that was used is NP_002284.3.
Results
Clinical findings
A Chinese family including two POI patients was enrolled in this study (). The 29-year-old proband (II-1) had undergone normal puberty, and menarche at 12 years of age. She had attended our hospital for the treatment of menopause for more than 3 months. Prior to this, she had experienced irregular menstruation more than one year, but no dysmenorrhea, headache, or other discomfort. There was no history of ovarian surgery, chemotherapy, radiotherapy, or immune disease.
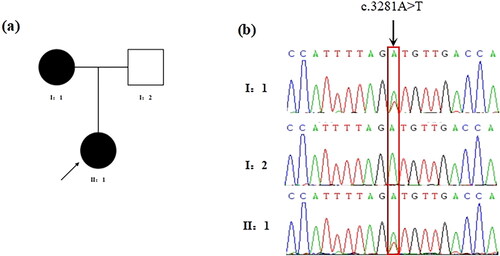
Figure 1. Identification of the missense variant of LAMC1 in the POI family. (a) Pedigree of the family with POI. Filled black symbols represent affected members. Arrow denotes the proband. (b) The chromatogram of the missense variant c.3281A > T (p.D1094V) of LAMC1 in the patient and his parents. The arrow denotes the variant site.
Physical examination showed a normal body mass index. FSH levels on two separate occasions >4 weeks apart were 29.15 and 25.62 IU/L, respectively (). Antral follicle counting on the third day of menstruation revealed no follicles in either ovary. Ultrasound examination showed the uterus to be 4.0 × 4.2 × 3.6 cm, an endometrial thickness of 0.35 cm, and right and left ovary sizes of 2.6 × 0.9 × 1.2 cm and 2.2 × 1.2 × 0.9 cm, respectively. There was no obvious follicular echo in either ovary.
Table 2. Hormonal characteristics of the proband.
Table 1. LAMC1-specific primers used for Sanger sequencing.
The proband’s mother started to have irregular menstrual cycles at the age of 34 years, and received HRT at the age of 38 years because of amenorrhea that had persisted for more than 3 months.
Identification of the missense LAMC1 variant
To investigate the genetic cause of POI in this family, we performed WES on the patient and her mother. Unrelated variants were excluded from sequencing results by rigorous bioinformatics analysis. In silico prediction revealed 50 deleterious variants, including six non-frameshift deletion variants, one frameshift deletion variant, two splicing variants, four nonsense variants, and 37 missense variants (Supplementary Table 1).
One of the novel missense variants was of LAMC1, which was previously identified as a gene associated with POI [Citation26]. Sanger sequencing confirmed that the proband and her mother carried the same missense variant (c.3281A > T, p.D1094V, ).
In silico analysis of the missense LAMC1 variant
The missense LAMC1 variant was shown to be located within domains I and II of the protein (). The minor allele frequency (MAF) of the variant was assessed in East Asian and global populations using the GnomAD database. Next, the impact of the variant on protein function was predicted by various bioinformatic tools. As shown on , the variant is absent from both East Asian and global populations. It was shown to be damaging by Sorting Intolerant from Tolerant (SIFT), possibly damaging by Polymorphism Phenotyping v2 (PolyPhen-2), and disease-causing by MutationTaster. Its Combined Annotation-Dependent Depletion (CADD) score was 26.5, the predict results of other tools were shown in Supplementary Table 2. And its American College of Medical Genetics (ACMG) classification was a variant of unknown significance (PM2, PP3). In addition, the LAMC1 variant was found to be strongly conserved among species (). Furthermore, Analysis of protein stability for the missense variant predicted a decrease in stability of the LAMC1 protein structure by I-Mutant2.0 and INPS-MD with free energy values of −0.60 and −0.045 Kcal/mol, respectively (Supplementary Table 2). Above results suggested a potentially pathogenicity of this missense variant and we hypothesized that this variant of LAMC1 might be the cause of this POI family.
Figure 2. Bioinformatics analysis of the identified missense variant of LAMC1. (a) The location of the variant in the intron-exon structure of LAMC1 and the protein domain map of LAMC1. The missense variant (c.3281A > T, p.D1094V) of LAMC1 is located in the domain I and II. (b) Amino acid alignment of the LAMC1 protein from several organisms. The position of Asp1094 residue (highlighted by a black box) was highly conserved among different species.
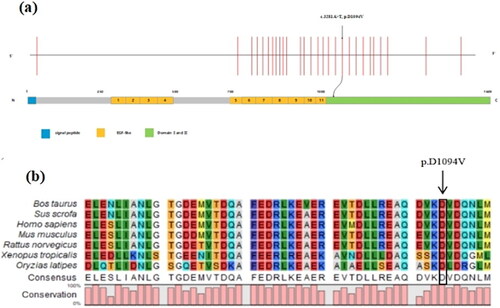
Table 3. Biological analysis of the LAMC1 c.3281A > T variant.
Discussion
In this study, we investigated the genetic cause of POI in a Chinese family with two affected females. The filtering of variants obtained by WES identified a novel variant of LAMC1, which was potentially causative of POI. Our study further provides evidence that LAMC1 variants are important in the pathogenesis of POI.
Laminins are a family of extracellular matrix proteins that are associated with the development of ovarian follicles [Citation27,Citation28]. They play an important role in cell adhesion, differentiation, migration, and signaling through interactions with integrins, dystroglycan, and other proteins [Citation29]. Mature laminins are crucial components of the basal lamina; together with collagens they affect the structure and function of numerous tissues [Citation30].
Laminins consist of three identical chains: laminin alpha, beta, and gamma (formerly A, B1, and B2, respectively). Each laminin chain is a multidomain protein encoded by a distinct gene which shows tissue-specific expression that changes during differentiation, resulting in the expression of specific mature laminin tripeptides in different tissues and cell types. Previous immunostaining studies have shown that some laminin chains, including the γ1 chain, are expressed in the basal lamina, and granulosa and/or theca cells of ovarian follicles in various animals [Citation31,Citation32], and in the human basal lamina around blood vessels and stroma, including theca interna cells of preovulatory follicles [Citation33]. Increased expression of laminins was documented in the bovine basal lamina during follicular development [Citation34], and similarly observed in pig and rabbit models [Citation32]. Laminins also affect the proliferation, survival, and steroidogenesis of human granulosa cells (GCs) in vitro, suggesting that they play a role in folliculogenesis [Citation35].
LAMC1 encodes the γ1 chain, which is the most abundantly expressed laminin subunit. LAMC1 is located on chromosome 1q25.3, contains 28 exons, and encodes a protein of 1609 amino acids that is expressed in a variety of human tissues, including the placenta, lung, heart, breast, and ovaries. According to the STRING database, LAMC1 might interacts with other laminin subunits such as LAMA3, LAMA5, and LAMB2 to affect laminin protein structure and function.
Previous human studies reported that LAMC1 was mainly expressed in follicular GCs, and that laminin tripeptides containing γ1 increased follicular survival in vitro [Citation36]. LAMC1 overexpression was found to enhance tumor cell invasion and migration and to predict poor ovarian cancer prognosis [Citation37], while LAMC1 polymorphisms were associated with an increased risk of developing pelvic organ prolapse and POI [Citation26,Citation38]. Furthermore, absence of the laminin γ1 chain by lamc1 deletion in mouse models led to aberrant basal lamina formation and embryonic lethality through the prevention of entire laminin polymerization [Citation39]. Laminin-null Schwann cells also exhibited the depletion of all other laminin chains, together with reduced phosphatidylinositol 3 kinase activity and activation of caspase cascades, leading to cell apoptosis [Citation40,Citation41]. We speculate that the disruption of LAMC1 causes POI by similarly preventing polymerization of the entire laminin molecule, resulting in the abnormal regulation of GC survival, which has a major role in follicular atresia.
In a recent genome-wide association study, Pyun et al. [Citation26] found that 22 single nucleotide polymorphisms (SNPs) of LAMC1 formed a linkage disequilibrium block in POI patients. This study showed that possession of at least one LAMC1 haplotype (C-C-T-G-C-C-A-T-T-C) and nine LAMC1 SNPs were associated with susceptibility to POI in the dominant model, suggesting that LAMC1 is involved in POI pathogenesis. This genetic model is consistent with that of the family in our study. However, there have been no previous reports about rare and potentially pathogenic variants of LAMC1 in POI patients, and especially in familial POI cases. Here, we used WES to identify a novel missense variant (c.3281A > T, p.D1094V) in LAMC1 in the proband and her affected mother of a Chinese POI family. The previously reported SNP (rs20558) is near the nidogen binding site γ1 III4, suggesting may interfere with the laminin–nidogen interaction, which is important for basal lamina stabilization [Citation42]. In our study, the mutated site is located in exon 19 of LAMC1, within domains I and II of the protein which are thought to interact with other laminin chains to form a coiled–coil structure serving a mechanical role in forming stiff bundles of fibers according to the uniprot database. Our findings suggested LAMC1 gene abnormality may affect basal lamina stabilization and thus follicle development through different biological processes. Furthermore, the position of this variant is highly conserved among mammals, and it was predicted to be potentially destructive to the structure and function of LAMC1 protein in in silico analysis.
In summary, the present study identified a novel missense variant (c.3281A > T, p.D1094V) of LAMC1 in a Chinese family with POI, which was inherited in an autosomal dominant manner. We speculate that this variant is causative of POI, and our findings suggest the importance of WES in the early detection and intervention of POI. However, the specific mechanism of the effect of this missense variant on laminin protein function is still unclear, further studies are required to clarify the association between the variant and POI through functional experiments, and larger sample sizes are needed to explore other genetic causes of POI.
Authors’ contributions
All authors contributed to the study conception and design. HFX and YGF collected the clinical samples and performed the clinical diagnosis. BBW, CYW and HW analyzed WES data. The validation experiment was performed by TYL. The first draft of the manuscript was written by CYW and HFX. All authors reviewed the manuscript.
Supplemental Material
Download MS Word (20.6 KB)Acknowledgements
We would like to acknowledge all participants in this study.
Disclosure statement
The authors declare that they have no competing interests.
Data availability statement
All data used during the study are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Chon SJ, Umair Z, Yoon MS. Premature ovarian insufficiency: past, present, and future. Front Cell Dev Biol. 2021;9:1. doi: 10.3389/fcell.2021.672890.
- McGlacken-Byrne SM, Conway GS. Premature ovarian insufficiency. Best Pract Res Clin Obstet Gynaecol. 2022 Nov;81:98–6. doi: 10.1016/j.bpobgyn.2021.09.011.
- Huang QY, Chen SR, Chen JM, et al. Therapeutic options for premature ovarian insufficiency: an updated review. Reprod Biol Endocrinol. 2022 Feb;20(1):28. doi: 10.1186/s12958-022-00892-8.
- Ishizuka B. Current understanding of the etiology, symptomatology, and treatment options in premature ovarian insufficiency (POI). Front Endocrinol (Lausanne). 2021;12:626924. doi: 10.3389/fendo.2021.626924.
- Jiao X, Ke H, Qin Y, et al. Molecular genetics of premature ovarian insufficiency. Trends Endocrinol Metab. 2018 Nov;29(11):795–807. doi: 10.1016/j.tem.2018.07.002.
- Liu H, Wei X, Sha Y, et al. Whole-exome sequencing in patients with premature ovarian insufficiency: early detection and early intervention. J Ovarian Res. 2020 Sep;13(1):114. doi: 10.1186/s13048-020-00716-6.
- He WB, Tan C, Zhang YX, et al. Homozygous variants in SYCP2L cause premature ovarian insufficiency. J Med Genet. 2021 Mar;58(3):168–172. doi: 10.1136/jmedgenet-2019-106789.
- Chen B, Li L, Wang J, et al. Consanguineous familial study revealed biallelic FIGLA mutation associated with premature ovarian insufficiency. J Ovarian Res. 2018 Jun;1811(1):48. doi: 10.1186/s13048-018-0413-0.
- Zhang YX, He WB, Xiao WJ, et al. Novel loss-of-function mutation in MCM8 causes premature ovarian insufficiency. Mol Genet Genomic Med. 2020 Apr;8(4):e1165. doi: 10.1002/mgg3.1165.
- Chen Q, Ke H, Luo X, et al. Rare deleterious BUB1B variants induce premature ovarian insufficiency and early menopause. Hum Mol Genet. 2020 Sep;29(16):2698–2707. doi: 10.1093/hmg/ddaa153.
- Jaillard S, McElreavy K, Robevska G, et al. STAG3 homozygous missense variant causes primary ovarian insufficiency and male non-obstructive azoospermia. Mol Hum Reprod. 2020 Sep;26(9):665–677. doi: 10.1093/molehr/gaaa050.
- Moriwaki M, Moore B, Mosbruger T, et al. POLR2C mutations are associated with primary ovarian insufficiency in women. J Endocr Soc. 2017 Mar;1(3):162–173. doi: 10.1210/js.2016-1014.
- Peng T, Lv C, Tan H, et al. Novel PMM2 missense mutation in a Chinese family with non-syndromic premature ovarian insufficiency. J Assist Reprod Genet. 2020 Feb;37(2):443–450. doi: 10.1007/s10815-019-01675-8.
- Caburet S, Todeschini AL, Petrillo C, et al. A truncating MEIOB mutation responsible for familial primary ovarian insufficiency abolishes its interaction with its partner SPATA22 and their recruitment to DNA double-strand breaks. EBioMedicine. 2019 Apr;42:524–531. doi: 10.1016/j.ebiom.2019.03.075.
- Chen A, Tiosano D, Guran T, et al. Mutations in the mitochondrial ribosomal protein MRPS22 lead to primary ovarian insufficiency. Hum Mol Genet. 2018 Jun;127(11):1913–1926. doi: 10.1093/hmg/ddy098.
- Heddar A, Guichoux N, Auger N, et al. A SPIDR homozygous nonsense pathogenic variant in isolated primary ovarian insufficiency with chromosomal instability. Clin Genet. 2022 Feb;101(2):242–246. doi: 10.1111/cge.14080.
- Fauchereau F, Shalev S, Chervinsky E, et al. A non-sense MCM9 mutation in a familial case of primary ovarian insufficiency. Clin Genet. 2016 May;89(5):603–607. doi: 10.1111/cge.12736.
- Sousa BL, Nishi MY, Santos MG, et al. Mutation analysis of NANOS3 in Brazilian women with primary ovarian failure. Clinics (Sao Paulo). 2016 Dec;71(12):695–698. doi: 10.6061/clinics/2016(12)03.
- Yang X, Touraine P, Desai S, et al. Gene variants identified by whole-exome sequencing in 33 french women with premature ovarian insufficiency. J Assist Reprod Genet. 2019 Jan;36(1):39–45. doi: 10.1007/s10815-018-1349-4.
- Wang Q, Li D, Cai B, et al. Whole-exome sequencing reveals SALL4 variants in premature ovarian insufficiency: an update on genotype-phenotype correlations. Hum Genet. 2019 Jan;138(1):83–92. doi: 10.1007/s00439-018-1962-4.
- Grzechocińska B, Warzecha D, Wypchło M, et al. Premature ovarian insufficiency as a variable feature of blepharophimosis, ptosis, and epicanthus inversus syndrome associated with c.223C > T p.(Leu75Phe) FOXL2 mutation: a case report. BMC Med Genet. 2019 Jul;20(1):132. doi: 10.1186/s12881-019-0865-0.
- Webber L, Davies M, Anderson R, European Society for Human R, Embryology Guideline Group on POI, et al. ESHRE guideline: management of women with premature ovarian insufficiency. Hum Reprod. 2016 May;31(5):926–937. doi: 10.1093/humrep/dew027.
- Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014 Mar;46(3):310–315. doi: 10.1038/ng.2892.
- Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005 Jul;33(Web Server issue):W306–W310. doi: 10.1093/nar/gki375.
- Savojardo C, Fariselli P, Martelli PL, et al. INPS-MD: a web server to predict stability of protein variants from sequence and structure. Bioinformatics. 2016 Aug;32(16):2542–2544. doi: 10.1093/bioinformatics/btw192.
- Pyun JA, Cha DH, Kwack K. LAMC1 gene is associated with premature ovarian failure. Maturitas. 2012 Apr;71(4):402–406. doi: 10.1016/j.maturitas.2012.01.011.
- Irving-Rodgers HF, Rodgers RJ. Extracellular matrix of the developing ovarian follicle. Semin Reprod Med. 2006 Sep;24(4):195–203. doi: 10.1055/s-2006-948549.
- Irving-Rodgers HF, Rodgers RJ. Extracellular matrix in ovarian follicular development and disease. Cell Tissue Res. 2005 Oct;322(1):89–98. doi: 10.1007/s00441-005-0042-y.
- Durbeej M. Laminins. Cell Tissue Res. 2010 Jan;339(1):259–268. doi: 10.1007/s00441-009-0838-2.
- Domogatskaya A, Rodin S, Tryggvason K. Functional diversity of laminins. Annu Rev Cell Dev Biol. 2012;28(1):523–553. doi: 10.1146/annurev-cellbio-101011-155750.
- Leardkamolkarn V, Abrahamson DR. Immunoelectron microscopic localization of laminin in rat ovarian follicles. Anat Rec. 1992 May;233(1):41–52. doi: 10.1002/ar.1092330107.
- Lee VH, Britt JH, Dunbar BS. Localization of laminin proteins during early follicular development in pig and rabbit ovaries. J Reprod Fertil. 1996 Sep;108(1):115–122. doi: 10.1530/jrf.0.1080115.
- Fujiwara H, Honda T, Ueda M, et al. Laminin suppresses progesterone production by human luteinizing granulosa cells via interaction with integrin alpha 6 beta 1. J Clin Endocrinol Metab. 1997 Jul;82(7):2122–2128. doi: 10.1210/jc.82.7.2122.
- Zhao Y, Luck MR. Gene expression and protein distribution of collagen, fibronectin and laminin in bovine follicles and corpora lutea. J Reprod Fertil. 1995 May;104(1):115–123. doi: 10.1530/jrf.0.1040115.
- Berkholtz CB, Shea LD, Woodruff TK. Extracellular matrix functions in follicle maturation. Semin Reprod Med. 2006 Sep;24(4):262–269. doi: 10.1055/s-2006-948555.
- Hao J, Tuck AR, Prakash CR, et al. Culture of human ovarian tissue in xeno-free conditions using laminin components of the human ovarian extracellular matrix. J Assist Reprod Genet. 2020 Sep;37(9):2137–2150. doi: 10.1007/s10815-020-01886-4.
- Diao B, Yang P. Comprehensive analysis of the expression and prognosis for laminin genes in ovarian cancer. Pathol Oncol Res POR. 2021;27:1609855.
- Chen J, Li L, Lang J, et al. Common variants in LAMC1 confer risk for pelvic organ prolapse in Chinese population. Hereditas. 2020 Jul;157(1):26. doi: 10.1186/s41065-020-00140-2.
- Smyth N, Vatansever HS, Murray P, et al. Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol. 1999 Jan;144(1):151–160. doi: 10.1083/jcb.144.1.151.
- Yu WM, Feltri ML, Wrabetz L, et al. Schwann cell-specific ablation of laminin gamma1 causes apoptosis and prevents proliferation. J Neurosci. 2005 May;25(18):4463–4472. doi: 10.1523/JNEUROSCI.5032-04.2005.
- McKee KK, Yang DH, Patel R, et al. Schwann cell myelination requires integration of laminin activities. J Cell Sci. 2012 Oct;125(Pt 19):4609–4619. doi: 10.1242/jcs.107995.
- Pöschl E, Fox JW, Block D, et al. Two non-contiguous regions contribute to nidogen binding to a single EGF-like motif of the laminin gamma 1 chain. Embo J. 1994;13(16):3741–3747. doi: 10.1002/j.1460-2075.1994.tb06683.x.