
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Objective
Estrogen (E2) is the main contributor to the progression of endometrial cancer (EC). The long noncoding RNA HOX antisense intergenic RNA (HOTAIR) is emerging as a new regulator in several cancer types. This study aimed to investigate the role of HOTAIR in EC development and identify the underlying molecular mechanisms.
Methods
HOTAIR expression levels in human EC tissues and the corresponding adjacent tissues and human EC Ishikawa cells were determined by quantitative PCR. Ishikawa cells were treated with E2 or estrogen receptor (ER) inhibitor ICI182780, transfected with siHOTAIR oligo, or infected with lentivirus expressing shHOTAIR/shNC, alone or in combinations. The protein expression of polycomb repressive complex 2 (PRC2) was evaluated by western blotting, and cell migration was measured by transwell assays. A xenograft tumorigenic model was established by inoculating control or stable shHOTAIR-infected Ishikawa cells into nude mice and implanting 17β-estradiol release pellets.
Results
HOTAIR expression was significantly elevated in human EC tissues. E2 exposure markedly increased HOTAIR levels in Ishikawa cells. Notably, E2 increased the protein expression of PRC2 and promoted EC cell migration, which were dependent on HOTAIR expression, as HOTAIR knockdown abolished these effects of E2. Similarly, E2 promoted the in vivo proliferation of grafted Ishikawa cells via upregulated HOTAIR expression in nude mice.
Conclusions
Human EC tissues highly express HOTAIR, and E2-induced EC progression depends on HOTAIR expression. This work suggests that the E2-HOTAIR axis is a potential therapeutic target in EC therapy.
Introduction
Endometrial cancer (EC) is one of the most commonly diagnosed gynecological cancers, accounting for 20%-30% of malignant tumors of the reproductive system in women [Citation1]. The 2022 cancer statistics found that the incidence of EC has increased significantly worldwide in recent years, showing a younger trend [Citation2,Citation3]. Estrogen-dependent (Type I) endometrial tumors, low grade endometrioid, make up the majority of EC cases (about 85%), are low grade with a glandular structure, usually express high levels of estrogen receptor α (ER), and are thought to be hormonally driven [Citation4,Citation5]. Since hormonally driven ECs are responsible for more deaths, the major risk factors and some treatment options for EC patients emphasize a key role for estrogen signaling in the disease [Citation6].
Estrogens play a mitogenic role in the normal endometrium, driving tissue growth as part of pregnancy anticipation during the menstrual cycle. In general, estrogens, including 17β-estradiol (E2), drive endometrial growth, and progestogens, including progesterone (P4), block endometrial growth and promote differentiation [Citation5]. During the late follicular phase of the menstrual cycle, the developing follicle produces estrogens, leading to the growth of the endometrium [Citation7]. In animal models, high levels of estrogens unopposed by progesterone lead to endometrial hyperplasia or cancer [Citation8,Citation9], suggesting that the lack of estrogen/progesterone balance can contribute to the early stages of EC formation. Although extensive studies have shown that excess unopposed estrogen exposure can increase the risk of EC [Citation5], the modulation mechanisms of estrogen in the development of EC remain poorly illustrated. Therefore, a deeper understanding focused on the functions of estrogen in EC occurrence and development is necessary.
Long noncoding RNAs (lncRNAs) are non-protein-coding RNAs crucial in genome regulation and major contributors to tumorigenesis [Citation10]. The lncRNA-HOX antisense intergenic RNA (HOTAIR) was the first type of lncRNA discovered by Rinn et al. [Citation11]. HOTAIR is 2.2 kb in length and localized on chromosome 12. Several cancer types overexpress HOTAIR, and HOTAIR is closely related to tumor cell proliferation and metastasis [Citation12]. HOTAIR can also promote the disease progression as competitive endogenous RNA [Citation13], activating signal transduction pathway [Citation14], participating in epithelial-mesenchymal transition [Citation15], and promoting protein ubiquitin [Citation16]. Notably, HOTAIR interacts with the polycomb repressive complex 2 (PRC2), and HOTAIR can increase the metastasis and invasiveness of breast cancer cells in a manner dependent on PRC2 [Citation17]. However, relatively few studies examined the roles of HOTAIR in EC, and the current reports are limited to the relationship between abnormal expression of HOTAIR and the malignant biological behavior of EC cells [Citation18–20]. In addition, the association between E2 exposure and expressions of HOTAIR and PRC2 in EC cells has not been explored.
This study aimed to investigate the role of HOTAIR in EC development and to identify the underlying molecular mechanisms. In this study, we first investigated the impact of estrogen exposure on HOTAIR expression in EC cell line Ishikawa and further examined whether estrogen can regulate PRC2 expression and migration in vitro and in vivo, which provides critical insight into developing novel therapeutic strategies for EC treatments.
Materials and methods
Endometrial cancer tissue samples
The EC tissue samples and the corresponding paracancerous tissue samples of 15 patients were obtained from the biological sample database of Beijing Obstetrics and Gynecology Hospital affiliated to Capital Medical University (Beijing, China). All patients were first diagnosed in this hospital and had no history of radiotherapy, chemotherapy, or other adjuvant therapy before surgery. All the specimens were immediately stored in liquid nitrogen until RNA extraction. The Ethical Committee of Beijing Obstetrics and Gynecology Hospital approved this study (Reference number: 2016-KY-061-01).
Cell culture and treatment
We purchased the human EC cell line Ishikawa from Alworson (Beijing, China). We maintained the Ishikawa cells in Roswell Park Memorial Institute (RPMI)-1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA), 100 U/mL of penicillin, and 100 μg/mL of streptomycin (Gibco, USA). We incubated the Ishikawa cells in a humidified atmosphere of 5% CO2 at 37 °C.
Before E2 treatment, we cultured the Ishikawa cells for at least three passages in phenol red-free RPMI-1640 medium (Gibco, USA) supplemented with 10% charcoal-stripped FBS (BI, Israel) to starve Ishikawa cells of steroid hormones. In order to investigate the effect of E2 on HOTAIR expression, we cultured the Ishikawa cells up to 60% confluence in 60-mm culture plates and treated them with various concentrations of E2 (Sigma-Aldrich, USA) for 4 h. Alternatively, we treated the cells with the optimal concentration of E2 for 0, 1, 2, 4, 6, 8 or 10 h.
Estrogen receptor (ER) antagonists are structurally similar to estrogen and inhibit the role of ER-mediated estrogen in tumor cells by competing with ER, which include tamoxifen and fulvestrant (ICI182780) and so on [Citation21]. Tamoxifen has dual regulatory effects. As an ER antagonist, tamoxifen is widely used in treating breast cancer, but it plays a weak estrogenic role in EC as an ER agonist to promote the progression of EC [Citation22]. Therefore, we used ICI182780, which only possesses an antagonistic effect on estrogen.
In some experiments, we treated the Ishikawa cells with the ER inhibitor ICI182780 (Selleck, USA), transfected them with siHOTAIR oligos (Sangon Biotech, China), or infected them with shHOTAIR-expressing lentivirus (Sangon Biotech, China), separately. After the indicated treatments and/or gene expression manipulation, we determined the expression levels of HOTAIR in these cells by quantitative PCR.
Small interfering RNA (siRNA)/short hairpin RNA (shRNA)-mediated HOTAIR knockdown
For in vitro cellular assays, we transfected the Ishikawa cells with 100 nM of siRNA targeting HOTAIR (GenBank NR_003716.3; NR_047517.1; NR-047518.1) using Lipofectamine™ 3000 transfection reagent (Invitrogen, USA) according to the manufacturer’s protocols. The siRNA oligo sequence was as follows: siHOTAIR, 5′-GAACGGGAGUACAGAGAGAUU-3′. We purchased the oligo from Sangon Biotech (Shanghai, China).
For in vitro cellular assays and xenograft tumor model experiments, we infected Ishikawa cells with lentivirus expressing HOTAIR-targeting shRNA. Three HOTAIR interference candidate target sequences and one negative control (NC) sequence were designed by Sangon Biotech (Shanghai, China). These sequences were as follows: shHOTAIR-2379 (5’-CCGGGAACGGGAGTACAGAGAGAATCTCGAGA TTCTCTCTGTACTCCCGTTCTTTTTT-3”); shHOTAIR-2380 (5’-CCGGGCAGCAC AGAGCAACTCTATACTCGAGTATAGAGTTGCTCTGTG CTGCTTTTTTG-3”); shHOTAIR-2381 (5’-CCGGGGACTGGGA GGCGCTAATTAACTCGAGTTAATTA GCGCCTCCCAGTCCTTTTTTG-3”) and shNC (5′-TTCTCCGAACGTGTCACGT-3”). The successful silencing of HOTAIR expression in Ishikawa cells was verified by qPCR.
Real-time-quantitative polymerase chain reaction (RT-qPCR)
We isolated the total RNA from frozen tissue specimens by homogenization in RiboEX reagent (GeneAll, Korea). TRIzol™ (Invitrogen, USA) was used to isolate total RNA from cells according to the manufacturer’s instructions. We used total RNA (1 µg) as a template to generate complementary DNA (cDNA) by oligonucleotides using the GoScript™ Reverse Transcription System (Promega, USA). Primer pairs were synthesized by Sangon Biotech (Shanghai, China) and used for qPCR. The sequences of the primers for qPCR were as follows: HOTAIR forward, 5′-GGTAGAAAAAGCAACCACGAAGC-3”′ and HOTAIR reverse, 5′-ACATAAACCTCTGTCTGTGAGTGCC-3′′; GAPDH forward, 5′-GCTGAGAACGGGAAGCTTGT-3” and GAPDH reverse, 5′-GCCAGGGGTGCTAAGCAG-3”. We conducted the qPCR experiment using Stratagene Mx3000P (Agilent Technologies, USA) in a final volume of 20 µL, which included 10 µL of GoTaq®qPCR Master Mix (Promega, USA), 0.4 µL of forward primer (10 µM), 1 µL of reverse primer, 1 µL of cDNA, and 7 µL of nuclease-free water. The PCR protocol was as follows: 94 °C for 30 s, followed by 45 cycles of 94 °C for 5s and 60 °C for 30s. We used GAPDH as a control for gene expression normalization.
Western blotting
We loaded 20 µg of each sample on a 7.5% polyacrylamide gel and transferred the proteins to polyvinylidene fluoride (PVDF) membranes by electroblotting. We blocked the membranes in 5% nonfat dry milk in Tris-buffered saline-Tween 20 (TBST). Primary antibody incubation was done using anti-SUZ12 (1:1000 dilution) and anti-EZH2 (1:500 dilution) antibodies (both purchased from Sigma, USA) overnight at 4 °C. The next day, we washed the blots four times in TBST and incubated them with horseradish peroxidase-conjugated goat anti-rabbit antibody (1:20,000 dilution) for 1 h at 37 °C. After washing again four times, we visualized the blots using an enhanced chemiluminescence system (Santa Cruz Biotechnology, USA) after incubation in the chemiluminescence reagent Femto™ (Thermo Scientific, USA) for 5 min. The light emitted was detected on a chemiluminescence detector (Tanon Science & Technology Co., Ltd., Shanghai, China). We analyzed the densitometry of each immunoblot using ImageJ (National Institutes of Health, USA), and protein expression was normalized to that of β-actin.
Transwell assay
We assessed cell migration using a Transwell assay (Millipore, USA) with a pore size of 0.8 μm. We seeded 5 × 105 cells in a serum-free medium in the upper chamber (a conventional chamber for the migration assay and a Matrigel™-coated chamber for the invasion assay) and filled the lower chamber with a medium containing 10% FBS. After incubation for 24 h at 37 °C, we carefully removed the cells in the upper chamber with a cotton swab. The invading cells on the bottom surface of the membrane were fixed in methanol and stained with 1% crystal violet counted. We took images using a microscope (IX71-A21PH, OLYMPUS, JAPAN) and counted the migrated/invaded cells from three randomly selected visual fields.
Experimental animals
We purchased 28 BALB/c nude mice (female, 4-5 weeks old, 16-18 g in body weight) from Beijing HFK Bioscience Co. Ltd. (Beijing, China) and randomly assigned them to four groups (n = 7 per group). We divided the mice in each group into two cages: one cage with four mice and the other with three mice. The mice were housed at the specific pathogen-free (SPF) facility in the Institute of Laboratory Animals Science, Chinese Academy of Medical Sciences & Peking Union Medical College (CAMS & PUMC) at room temperature (22 ± 1 °C) with a 12/12-h light/dark cycle and access to normal diet and water ad libitum. The Animal Welfare Committee of Capital Medical University (AEEI-2018-196) approved all experiments involving the use of animals. The experiments were conducted in the Institute of Laboratory Animals Science, CAMS & PUMC.
Xenograft tumor model
Before inoculating the Ishikawa cells, we performed bilateral ovariectomy in nude mice to establish a model of estrogen deficiency to rule out the interference of endogenous estrogen in female nude mice. We implanted the 17β-estradiol 60-day release pellet (0.72 mg/pellet, Sigma, USA) subcutaneously into the neck of the three groups of nude mice that needed E2 treatment. We selected the dose of E2 according to a previous publication [Citation20]. We inoculated subcutaneously Control, shNC-expressing, and shHOTAIR-expressing Ishikawa cells (5 × 106/100 µL per mouse) in the logarithmic growth phase into the right lower back of nude mice in the designated groups. Every 3 days after inoculation, we recorded the tumor formation time, the tumor formation rate, and the growth curve of the xenograft tumor. The tumor volume (mm3) was calculated as follows: V = 1/2 × length × width2. The experiment was terminated on the 28th day after tumor inoculation (or when the volume of the transplanted tumor reached 500 mm3 for individual mice). All mice showed unruptured tumors at the time of sacrifice. We completely removed the tumors and measured their volume and weight.
Statistical analyses
We conducted the statistical analyses using SPSS 23.0 (IBM, USA) and GraphPad Prism 7.0 (GraphPad Software, USA). We repeated all experiments in triplicate. If the experimental data accorded with normal distribution, all values are expressed by mean ± standard deviation ( ± s). We compared the variance between groups using the student’s t-test or the one-way analysis of variance (ANOVA). A p < 0.05 was considered statistically significant.
Results
Human EC tissues have a higher expression level of HOTAIR
To explore the potential relationship between HOTAIR expression and the development of EC, we performed RT-qPCR analyses using tumor tissues and the adjacent normal tissues resected from the same cohort of patients. Notably, compared with the corresponding control normal tissues, EC tumors had prominently upregulated HOTAIR expression (p < 0.01; ). Therefore, EC tissues have increased transcript levels of HOTAIR, suggesting that HOTAIR may be involved in the development of EC.
Figure 1. E2 upregulated HOTAIR expression in Ishikawa cells. (a) The expression of HOTAIR in human endometrial cancer tissue (tumor) and the adjacent normal tissue (control) from endometrial cancer (EC) patients was determined by quantitative polymerase chain reaction (qPCR). ** P<0.01, n = 15 for each group. (b,c) The expression of HOTAIR in Ishikawa cells treated with various concentrations of estradiol (E2) (b), or treated with 10 nM E2 for 0, 1, 2, 4, 6, 8, or 10 h (c), was determined by qPCR. (d) The expression of HOTAIR in Ishikawa cells left untreated, treated with E2 alone, or treated with E2 plus estrogen inhibitor ICI 182780, was determined by qPCR. (e-f) The expression of HOTAIR in control Ishikawa cells and cells transfected with small interfering RNA against HOTAIR (siHOTAIR) (e) or infected with lentivirus expressing small hairpin RNA (shRNA) against HOTAIR (shHOTAIR) or a negative control shRNA (shNC) (f), was determined by qPCR. (g) Control Ishikawa cells and the cells infected with shNC or shHOTAIR-expressing lentivirus were treated with 10 nM E2 for 4 h. The expression of HOTAIR in the indicated 4 groups was determined by qPCR. *** P<0.001, n = 3 for each group.
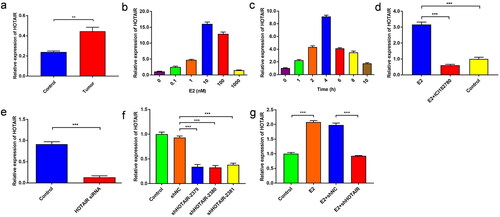
E2 increases HOTAIR expression in Ishikawa cells
We evaluated the impacts of E2 treatments on the expression of HOTAIR in Ishikawa cells. As shown in , E2 significantly upregulated the HOTAIR level in a dose-dependent manner. When exposed to 10 nM E2, Ishikawa cells showed increased expression of HOTAIR in a time-dependent manner (). In addition, additional treatment with the ER inhibitor ICI182780 abolished the remarkable E2-mediated increase in HOTAIR expression (), suggesting the specificity of E2/ER signaling in upregulating HOTAIR expression.
In order to examine the targeting specificity in HOTAIR for E2, HOTAIR expression was knocked down in Ishikawa cells. The results showed that the cells transfected with siRNA had a downregulated expression of HOTAIR (p < 0.001) (). The same was observed for the stable cell lines with lentivirus-mediated interference of HOTAIR expression () (p < 0.001). Also, the E2 group had significantly higher relative mRNA levels of HOTAIR compared with the control group (p < 0.001), while the E2 + shHOTAIR group had a significantly lower HOTAIR level than the E2 + shNC group (p < 0.001). However, the E2 + shHOTAIR group after E2 treatment did not show reduced HOTAIR expression than the control group (). These results suggest that E2 can specifically upregulate HOTAIR expression in Ishikawa cells.
E2 promotes PRC2 expression and migration of Ishikawa cells via upregulated HOTAIR expression
E2 significantly increased the protein expression of EZH2 and SUZ12, two core components of PRC2 (p < 0.001) (). However, siRNA-mediated targeted knockdown of HOTAIR prominently suppressed the upregulated expression of PRC2 (EZH2 and SUZ12) in Ishikawa cells (p < 0.001) (), which implies that E2 can increase PRC2 expression in Ishikawa cells via HOTAIR upregulation.
Figure 2. E2 promotes PRC2 expression and migration of Ishikawa cells via upregulated HOTAIR expression. (a–b) The protein expression of PRC2 (EZH2 and SUZ12) in Ishikawa cells treated with/without 10 nM estradiol (E2) for 4 h was determined by western blot assays. (c,d) The protein expression of PRC2 (EZH2 and SUZ12) in control Ishikawa cells and small interfering RNA against HOTAIR (siHOTAIR)-transfected Ishikawa cells after 10 nM E2 treatment for 4 h was determined by western blot assays. Representative band images are shown (a, c), and the relative protein levels were summarized (b,d). *** P<0.001, n = 3 for each group. (e–g) The migration capacity of Ishikawa cells was determined in the presence of E2 using the Transwell assay. The Ishikawa cells were treated without E2 (e) or with E2 (f). (h–j) The migration capacity of Ishikawa cells treated with E2 was determined with siHOTAIR-induced HOTAIR knockdown using the Transwell assay: without HOTAIR knockdown (h) and with HOTAIR-knockdown (i). Representative images of the Transwell assays are shown in e,f,h, and i, and the number of migrated cells per visual field was summarized (g,j). Magnification, 200 ×. *** P < 0.001, n = 3 for each group.
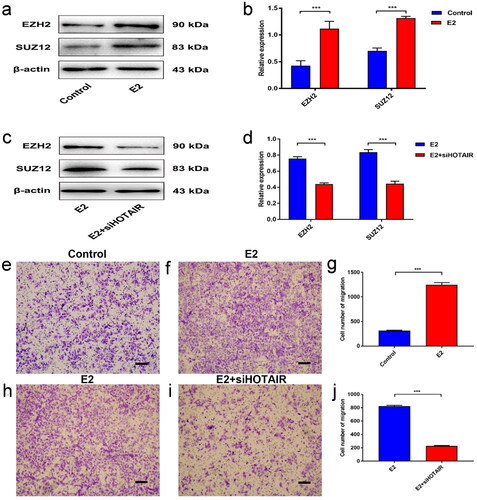
Moreover, to investigate the role of HOTAIR in EC metastasis, we determined the migration capacity in HOTAIR-silenced Ishikawa cells. In control cells, we found that the percentage of cells that traveled through the micropore membrane was significantly increased in E2-stimulated cells as compared to control (p < 0.001) (). However, HOTAIR-silenced cells displayed significantly suppressed migration in comparison to control cells upon E2 stimulation (p < 0.001) (). These results suggested that E2 can promote the migration of Ishikawa cells via HOTAIR upregulation.
E2 promotes the in vivo proliferation via upregulated HOTAIR
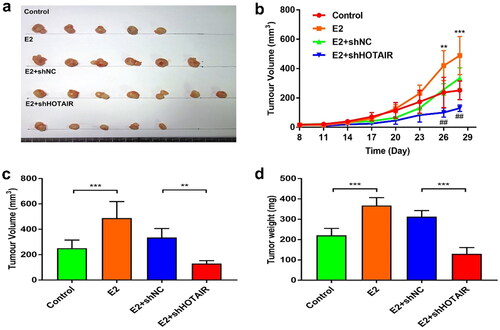
To further understand whether the role of E2 in tumor growth depended on HOTAIR expression, we established stable Ishikawa cell lines and inoculated them into ovariectomized nude mice. Nude mice in the E2 and E2 + shNC groups all formed tumors on the 8th day of EC inoculation, and the tumor formation rate was 100%. For unknown reasons, one mouse in the E2 group died on day 15 after cell inoculation. Only five mice in the control group and two mice in the E2 + shHOTAIR group (increased to five mice on the 11th day) showed tumor formation on day 8. The final tumor formation rates of these two groups were 71.4% (). In addition, the tumor growth curve confirmed that tumors formed from the E2 and E2 + shNC groups grew faster than the tumors formed from the control and E2 + shHOTAIR groups (E2 vs. Control, p < 0.001; E2 + shNC vs. E2 + shHOTAIR, p < 0.01) (). Moreover, the same trends regarding tumor weight were observed (E2 vs. Control, p < 0.001; E2 + shNC vs. E2 + shHOTAIR: p < 0.001) (). Thus, it appeared that E2 promoted the in vivo proliferation of Ishikawa cells in a HOTAIR-dependent manner.
Figure 3. E2-enhanced in vivo proliferation of Ishikawa cells was dependent on HOTAIR expression. (a–d) Nude mice were inoculated with control Ishikawa cells or stable Ishikawa cells with lentivirus infection-mediated expression of small hairpin RNA (shRNA) against HOTAIR (shHOTAIR) or a negative control shRNA (shNC) on Day 0. Mice in the estradiol (E2) treatment groups were subcutaneously implanted with 17β-estradiol-releasing pellets (a) Images of tumors at day 28 from the mice indicated in the different groups. (b) The growth curve of xenograft tumors in the indicated groups (*, E2 vs. Control; #, E2 + shNC vs. E2 + shHOTAIR). (c,d) Xenograft tumor volume (c) and weight (d) at the endpoint (day 28) were measured. ** P<0.01, *** P<0.001, ## P<0.01, n = 5 for all groups in b, c, d.
Discussion
Recently, multiple lncRNAs play important roles in the development and progression of EC [Citation23]. Here, we found that human EC tissues had a significantly upregulated expression of lncRNA HOTAIR compared with matched adjacent tissues, consistent with the report from Huang et al. [Citation18]. In addition, we showed that the HOTAIR knockdown markedly inhibited the expression of PRC2 (EZH2 and SUZ12) and the migration of the EC cell line Ishikawa. These results prompted us to hypothesize that HOTAIR might play a critical tumor-promoting role in EC progression.
EC (type I) originates from glandular epithelium, and excessive exposure to estrogen is an important pathogenic factor of EC [Citation24], but the specific mechanism of estrogen in promoting the development and progression of EC is not clear. A report from Bhan et al. showed that the transcription of HOTAIR in human breast cancer MCF7 cells (ER-positive) is positively regulated by estrogen [Citation25]. Our results showed that estrogen can induce HOTAIR expression in a dose- and time-dependent manner. There are some ERE sequences in the promoter region of HOTAIR, and estrogen can induce HOTAIR transcription by combining the ER to form a dimer and then binding to EREs on the HOTAIR promoter [Citation25]. Since the same effects of estrogen regarding upregulating HOTAIR expression were noticed in both breast cancer cells and EC cells, it is plausible that the same mechanism of estrogen might be applied to Ishikawa cells in our experimental settings. Interestingly, high levels of E2 inhibited HOTAIR expression. It is possible that low levels of E2 enhance HOTAIR expression but that high levels of E2 have an inhibitory effect on HOTAIR. The present study was not designed to determine the mechanisms responsible for those results, but they are interesting and will warrant further investigation.
The ER inhibitor ICI182780 [Citation21] remarkably abrogated estrogen’s ability to promote the expression of HOTAIR in Ishikawa cells, suggesting that estrogen down-regulated HOTAIR expression through the ER pathway. Moreover, in HOTAIR-knockdown Ishikawa cells, E2 still upregulated HOTAIR expression to a level comparable to that in control Ishikawa cells without E2 exposure, indicating that increased transcription but not by reduced degradation explained the E2-induced HOTAIR expression.
A recent study investigated the role of HOTAIR in molecular scaffolds [Citation17]. Notably, the classic process increases the metastasis and invasiveness of breast cancer cells in a manner dependent on PRC2 [Citation17]. PRC2 is a histone methyltransferase involved in epigenetic silencing during cancer development [Citation26,Citation27], and the EZH2 plays a major part in the transfer of methyl groups. Conversely, the finding of Manuela et al. suggests that forced overexpression of HOTAIR in breast cancer cells leads to subtle transcriptomic changes that appear independent of PRC2 [Citation28]. We found that E2 can increase the PRC2 (EZH2 and SUZ12) protein expression and migration of Ishikawa cells, which was further proven to be dependent on HOTAIR expression. These results confirmed that PRC2 functioned downstream of HOTAIR in Ishikawa cells, and E2 treatment may cause transcriptional induction of HOTAIR and subsequent increase in PRC2 expression.
Similar to what was observed in the in vitro experiments, HOTAIR-knockdown cells displayed significantly slowed growth than the control cells upon comparable doses of E2 exposure during the 28-day observation period, further strengthening the roles of upregulated HOTAIR expression in E2-induced EC progression. In addition, one tumor-bearing nude mouse died on the 15th day after inoculation in the E2 group. The reason might be due to the poor immune system of nude mice. Moreover, as estrogen can promote the growth of transplanted tumors through activating the phosphatidylinositol 3-kinase/Akt signaling pathway [Citation19], the too-rapid proliferation of tumors may affect the physiological function of multiple organs and lead to mouse death.
In conclusion, we reported here that human EC tissues highly express HOTAIR, and estrogen increases the expression of HOTAIR in EC cells. Moreover, estrogen upregulates PRC2 expression and promotes the migration of EC cells in vitro and the in vivo proliferation of EC cells via upregulated HOTAIR expression. Although the specific mechanism of estrogen in regulating HOTAIR expression in EC still needs to be further elucidated, our study provides a novel mechanistic insight into E2-associated EC pathogenesis. The expression of HOTAIR should be explored in multiple EC cell lines.
Authors’ contributions
Huixiao Wang and Ziwen Jiang conceived and designed the study. Huixiao Wang, Di Xia, and Ziwen Jiang performed the experiments. Di Xia analyzed the data and did the statistical analysis. Xulan Ma and Huixiao Wang wrote the paper. Yinmei Dai, Feng Sui, and Fengxian Fu reviewed and edited the manuscript. All authors read and approved the manuscript.
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki. This study was approved by the Ethical Committee of Beijing Obstetrics and Gynecology Hospital (Reference number: 2016-KY-061-01).
Disclosure statement
The authors report there are no competing interests to declare.
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Passarello K, Kurian S, Villanueva V. Endometrial cancer: an overview of pathophysiology, management, and care. Semin Oncol Nurs. 2019;35(2):1–7. doi: 10.1016/j.soncn.2019.02.002.
- Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33. doi: 10.3322/caac.21708.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442.
- Suarez AA, Felix AS, Cohn DE. Bokhman redux: endometrial cancer “types” in the 21st century. Gynecol Oncol. 2017;144(2):243–249. doi: 10.1016/j.ygyno.2016.12.010.
- Rodriguez AC, Blanchard Z, Maurer KA, et al. Estrogen signaling in endometrial cancer: a key oncogenic pathway with several open questions. Horm Cancer. 2019;10(2-3):51–63. doi: 10.1007/s12672-019-0358-9.
- Setiawan VW, Yang HP, Pike MC, et al. Type I and II endometrial cancers: have they different risk factors? J Clin Oncol. 2013;31(20):2607–2618. doi: 10.1200/JCO.2012.48.2596.
- Groothuis PG, Dassen HH, Romano A, et al. Estrogen and the endometrium: lessons learned from gene expression profiling in rodents and human. Hum Reprod Update. 2007;13(4):405–417. doi: 10.1093/humupd/dmm009.
- Yang CH, Almomen A, Wee YS, et al. An estrogen-induced endometrial hyperplasia mouse model recapitulating human disease progression and genetic aberrations. Cancer Med. 2015;4(7):1039–1050. doi: 10.1002/cam4.445.
- Vollmer G. Endometrial cancer: experimental models useful for studies on molecular aspects of endometrial cancer and carcinogenesis. Endocr Relat Cancer. 2003;10(1):23–42. doi: 10.1677/erc.0.0100023.
- Gutschner T, Diederichs S. The hallmarks of cancer: a long noncoding RNA point of view. RNA Biol. 2012;9(6):703–719. doi: 10.4161/rna.20481.
- Rinn JL, Kertesz M, Wang JK, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129(7):1311–1323. doi: 10.1016/j.cell.2007.05.022.
- Yuan C, Ning Y, Pan Y. Emerging roles of HOTAIR in human cancer. J Cell Biochem. 2020;121(5-6):3235–3247. doi: 10.1002/jcb.29591.
- Liu Y, Chen X, Chen X, et al. Long noncoding RNA HOTAIR knockdown enhances radiosensitivity through regulating microRNA-93/ATG12 axis in colorectal cancer. Cell Death Dis. 2020;11(3):175. doi: 10.1038/s41419-020-2268-8.
- Salmeron-Barcenas EG, Illades-Aguiar B, Del Moral-Hernandez O, et al. HOTAIR knockdown decreased the activity wnt/beta-Catenin signaling pathway and increased the mRNA levels of its negative regulators in hela cells. Cell Physiol Biochem. 2019;53(6):948–960.
- Song Y, Wang R, Li LW, et al. Long noncoding RNA HOTAIR mediates the switching of histone H3 lysine 27 acetylation to methylation to promote epithelial-to-mesenchymal transition in gastric cancer. Int J Oncol. 2019;54(1):77–86. doi: 10.3892/ijo.2018.4625.
- Xue M, Chen LY, Wang WJ, et al. HOTAIR induces the ubiquitination of Runx3 by interacting with Mex3b and enhances the invasion of gastric cancer cells. Gastric Cancer. 2018;21(5):756–764. doi: 10.1007/s10120-018-0801-6.
- Gupta RA, Shah N, Wang KC, et al. Long noncoding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–1076. doi: 10.1038/nature08975.
- Huang J, Ke P, Guo L, et al. Lentivirus-mediated RNA interference targeting the long noncoding RNA HOTAIR inhibits proliferation and invasion of endometrial carcinoma cells in vitro and in vivo. Int J Gynecol Cancer. 2014;24(4):635–642. doi: 10.1097/IGC.0000000000000121.
- Zhang XH, Hu P, Xie YQ, et al. Long noncoding RNA HOTAIR promotes endometrial carcinoma cell proliferation by binding to PTEN via the activating phosphatidylinositol 3-Kinase/akt signaling pathway. Mol Cell Biol. 2019;39(23):e00251–19. doi: 10.1128/MCB.00251-19.
- Zhou YX, Wang C, Mao LW, et al. Long noncoding RNA HOTAIR mediates the estrogen-induced metastasis of endometrial cancer cells via the miR-646/NPM1 axis. Am J Physiol Cell Physiol. 2018;314(6):C690–C701. doi: 10.1152/ajpcell.00222.2017.
- Sharma D, Kumar S, Narasimhan B. Estrogen alpha receptor antagonists for the treatment of breast cancer: a review. Chem Cent J. 2018;12(1):107. doi: 10.1186/s13065-018-0472-8.
- Janacova L, Faktor J, Capkova L, et al. SWATH-MS analysis of FFPE tissues identifies stathmin as a potential marker of endometrial cancer in patients exposed to tamoxifen. J Proteome Res. 2020;19(7):2617–2630. doi: 10.1021/acs.jproteome.0c00064.
- Li BL, Wan XP. The role of lncRNAs in the development of endometrial carcinoma. Oncol Lett. 2018;16(3):3424–3429. doi: 10.3892/ol.2018.9065.
- Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol. 1983;15(1):10–17. doi: 10.1016/0090-8258(83)90111-7.
- Bhan A, Hussain I, Ansari KI, et al. Antisense transcript long noncoding RNA (lncRNA) HOTAIR is transcriptionally induced by estradiol. J Mol Biol. 2013;425(19):3707–3722. doi: 10.1016/j.jmb.2013.01.022.
- Liu X, Liu X. PRC2, chromatin regulation, and human disease: insights from molecular structure and function. Front Oncol. 2022;12:894585. doi: 10.3389/fonc.2022.894585.
- Fang X, Ni N, Wang X, et al. EZH2 and endometrial cancer development: insights from a mouse model. Cells. 2022;11(5):909. doi: 10.3390/cells11050909.
- Portoso M, Ragazzini R, Brencic Z, et al. PRC2 is dispensable for HOTAIR-mediated transcriptional repression. Embo J. 2017;36(8):981–994. doi: 10.15252/embj.201695335.