
Abstract
Platelets may play a role in the high risk for vascular complications in Gram-positive sepsis. We compared the platelet reactivity of 15 patients with Gram-positive sepsis, 17 with Gram-negative sepsis and 20 healthy controls using a whole blood flow cytometry-based assay. Patients with Gram-positive sepsis had the highest median fluorescence intensity (MFI) of the platelet membrane expression of P-selectin upon stimulation with high dose adenosine diphosphate (ADP; P = 0.002 vs. Gram-negative and P = 0.005 vs. control groups) and cross-linked collagen-related peptide (CRP-XL; P = 0.02 vs. Gram-negative and P = 0.0001 vs. control groups). The Gram-positive group also demonstrated significantly higher ADP-induced fibrinogen binding (P = 0.001), as wll as platelet-monocyte complex formation (P = 0.02), compared to the Gram-negative group and had the highest plasma levels of platelet factor 4, β-thromboglobulin and soluble P-selectin. In contrast, thrombin-antithrombin complex and C-reactive protein levels were comparable in both patient groups. In conclusion, common Gram-positive pathogens induce platelet hyperreactivity, which may contribute to a higher risk for vascular complications
Introduction
The concept that platelets play an important role in immunity and host defence, besides their role in haemostasis, has become widely accepted. Many different bacteria, viruses and other micro-organisms have been shown to interact with platelets, changing the platelet phenotype and the interaction of platelets with leukocytes [Citation1,Citation2]. Whereas platelet activation is important in innate immune responses, excessive platelet activation may contribute to organ dysfunction and increase the risk for cardiovascular events (CVE) and infective endocarditis [Citation3,Citation4]. Acute bacterial infections are associated with an increased risk for CVE, but the magnitude of this risk, as well as the risk for infective endocarditis, depends on the type of infections [Citation5–Citation7].
Bacteria are generally divided into Gram-positive and Gram-negative bacteria. In contrast to Gram-negative bacteria, Gram-positive bacteria usually have a thick multi-layered peptidoglycan cell wall rich in teichoic acid with virtually absent lipopolysaccharide (LPS) [Citation8]. Clinical studies have shown a high incidence of cardiovascular complications in patients with Gram-positive bacterial infection [Citation9,Citation10]. This risk appears lower for urinary tract infections, which are usually caused by Escherichia coli or other Gram-negative bacteria [Citation5,Citation11]. In addition, infective endocarditis is predominantly caused by Gram-positive bacteria [Citation12].
Laboratory studies have shown that Gram-positive bacteria, such as Staphylococcus aureus and Streptococcus spp., bind to and activate platelets, whereas this has not been demonstrated for most Gram-negative bacteria [Citation13]. Interestingly, our group recently demonstrated that bacteremia due to Streptococcus pneumoniae in pigs leads to platelet hyperreactivity [Citation14]. On the other hand, data on the platelet-activating properties of E. coli LPS, which exerts potent inflammatory effects, have been conflicting and challenging to translate to the human in vivo setting [Citation15–Citation18].
Studies on the platelet-activating properties of bacteria were most frequently done either ex vivo or in animal models. To the best of our knowledge, no study has provided human in vivo data comparing platelet reactivity in patients with Gram-positive or Gram-negative sepsis. We therefore studied platelet reactivity, platelet-monocyte complex (PMC) formation and activation of the plasmatic coagulation in a cohort of patients with either Gram-positive or Gram-negative sepsis.
Methods
Study population
We enrolled adult patients admitted with a Gram-positive or Gram-negative sepsis in the Radboud university medical centre, Nijmegen, The Netherlands. A list of patients with positive blood cultures was generated daily by the Department of Medical Microbiology, and patients were reviewed for inclusion and exclusion criteria. Blood was drawn for platelet reactivity and haemostasis tests within 72 hours after the first blood culture collection and initiation of antibiotic treatment. Exclusion criteria were admission to the intensive care unit (ICU), renal replacement therapy, use of P2Y12 receptor antagonists, active malignancy, the presence of any chronic viral infection, including HIV or hepatitis B/C and a positive blood culture with a possible contaminant (e.g. coagulase-negative staphylococci). ICU patients were deliberately excluded to avoid confounding of organ failure on the assessment of the effects of bacteremia on platelet reactivity. Use of a low dose of aspirin or vitamin K antagonists was not a reason for exclusion, as these drugs have no influence on the platelet reactivity assay used [Citation19,Citation20]. A group of healthy volunteers were enrolled as controls. Patients and healthy controls were included after written informed consent was obtained. The study is approved by the ethical committee of the Radboud university medical centre.
Laboratory assays
Platelet reactivity
Venous blood was collected in citrated Vacutainer tubes (3.2% sodium citrate; Becton Dickinson, USA). Platelet reactivity was determined within 1 hour after blood drawing by a flow cytometry assay that was described earlier [Citation21,Citation22]. In short, the expression of the α-granule protein P-selectin and binding of fibrinogen to the activated integrin αIIbβ3 are measured as markers of platelet degranulation and aggregation, respectively, in unstimulated samples and after ex vivo platelet stimulation by adenosine diphosphate (ADP, low dose of 7.8 μM and high dose of 31.2 μM, Sigma-Aldrich, USA) or cross-linked collagen-related peptide (CRP-XL, low dose of 39 ng/L and high dose of 625 ng/L, kind gift from Prof. dr. R. Farndale, Cambridge, UK). Whole blood was added to a mixture of HEPES-buffered saline and saturating concentrations of PE-labelled anti-CD62P (P-selectin; Bio-Legend, San Diego, USA), FITC-labelled anti-fibrinogen (DAKO Ltd., High Wycombe, UK) and PC7-labelled anti-CD61 (platelet identification marker; Beckman Coulter, France). After 20 minutes incubation at room temperature, 0.2% paraformaldehyde was added and samples were analysed using an FC500 flow cytometer (Beckman Coulter, France). Platelets were gated based on their forward- and sideward-scatter properties and positivity for CD61, which was defined as a median fluorescence intensity (MFI) exceeding that of its matched isotype control. Next, the MFI of CD62P and fibrinogen relative to their matched isotype controls on CD61-positive events was determined.
Platelet-monocyte complexes
The formation of PMC, which is considered a sensitive marker for platelet activation [Citation23], was determined by incubating citrated whole blood with PC7-labelled anti-CD61 and PE-labelled anti-CD14 [a glycosylphosphatidylinositol (GPI)-linked membrane glycoprotein; Bio-Legend] as a monocyte identification marker. Optilyse B, which contains both lysing buffer and fixative (Beckman Coulter, USA), was added after 30 min followed by distilled water. The PMC formation was quantified based on the MFI of CD61 on CD14-positive cells.
Soluble platelet proteins and thrombin-antithrombin complexes
Platelet-poor plasma was harvested from citrate anticoagulated whole blood by centrifugation (1500 g without brake, 15 min, 20°C). Plasma concentrations of platelet factor 4 (PF4), β-thromboglobulin (β-TG), soluble P-selectin and thrombin-antithrombin (TAT) complexes were subsequently measured using ELISA as previously described [Citation24]. Human PF4 (MAB7951, AF795), β-thromboglobulin (MAB393, BAF393) and soluble P-selectin (DYE137) antibodies were purchased form R&D systems, Abbington, UK. Sheep anti-human thrombin (SAHT-AP, SAHT-HRP) antibodies were purchased from Kordia/Affinity Biologicals, USA.
Full blood count and C-reactive protein
A full blood count was determined using a standard haematology analyser (Sysmex XE 5000) calibrated for standard patient care. C-reactive protein (CRP) was determined using immunologic agglutination detection, and the urinary concentration of creatinine was determined with enzymatic colorimetric detection using Abbott Aeroset analyser (Abbott Laboratories).
Statistical analysis
Differences in patient characteristics across groups were compared using analysis of variance (ANOVA) with post-tests and chi-square test for proportions. Data on platelet reactivity are expressed as medians with interquartile range (IQR), while data on plasma soluble markers and PMC formation are expressed as medians with IQR, minimum and maximum values. The Mann–Witney U test was used to analyse statistical differences in the time intervals between the haemostatic tests and antibiotic treatment initiation or blood culture, in the CRP levels among the patient groups and in the platelet reactivity of patients with or without aspirin. Platelet reactivity and plasma markers levels between multiple groups were compared using the non-parametric Kruskal–Wallis test with Dunn’s post-test. Analyses were performed with GraphPad Prism (GraphPad Software, USA). P values less than 0.05 were considered statistically significant.
Results
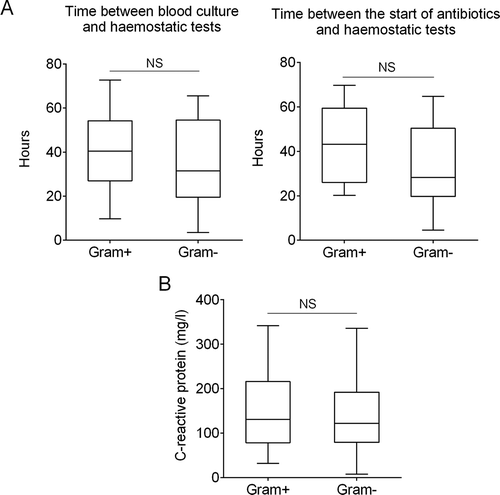
Thirty-two patients with sepsis were enrolled, of whom 15 had a Gram-positive and 17 a Gram-negative sepsis, with 20 healthy individuals as controls (). Patients in the Gram-positive group presented clinically with sepsis (n = 8), erysipelas (n = 3), pneumonia (n = 2) and infected diabetic foot ulcer (n = 1). The most common clinical diagnosis in the Gram-negative group was urosepsis (n = 11), followed by cholecystitis and intravascular catheter-related infection (each n = 2), as well as endometritis and infected diabetic foot ulcer (each n = 1). Patient characteristics in both groups were similar, including time intervals between the haemostatic tests and antibiotic treatment initiation or blood culture (). The levels of CRP, measured on the day (±24 hrs) when haemostasis assays were also performed, were comparable across both patient groups (). The mean age of the controls was lower than the patients in both sepsis groups.
Table I. Characteristics of study population.
Figure 1. Timing of haemostatic tests and plasma concentration of C-reactive protein (CRP). Box plots of the (A) time interval between platelet reactivity and haemostatic test and positive blood culture (left panel) or start of antibiotics (right panel). (B) CRP levels at the day (+/- 24 hrs) of the haemostasis assays. Presented data are medians with IQR, minimum and maximum values. NS, not statistically significant.
Participants in the Gram-positive group had a significantly higher ADP- and CRP-XL-induced P-selectin expression than participants in the Gram-negative group and controls (). Upon stimulation with high dose ADP (31.2 uM), the Gram-positive group demonstrated the highest MFI of P-selectin (median 49.2, IQR 34.6–55.2) compared to the Gram-negative (28.1, 25.3–38.3; P = 0.002) and control groups (29.7, 26.2–35.7; P = 0.005). When induced with high dose CRP-XL (625 ng/ml), patients in the Gram-positive group had a significantly higher P-selectin (89.8, 72.4–95.4) than both the Gram-negative (69, 67.7–80.3; P = 0.02) and control groups (67.6, 64.3–72.4; P = 0.0001). ADP-induced platelet-fibrinogen binding was also significantly higher in this group (18.3, 15.6–24.2 vs. 12.6, 10.8–13.9; P = 0.001 vs. Gram-negative group). There were no differences in these parameters between the Gram-negative group and the controls.
Figure 2. Platelet reactivity and platelet-monocyte complex in patients with Gram-positive and Gram-negative sepsis. (A) Platelet membrane expression of P-selectin and platelet-fibrinogen binding is depicted as median fluorescence intensity (MFI) in arbitrary units, at baseline and after stimulation with two concentrations of the platelet agonists adenosine diphosphate (ADP) and collagen-related peptide (CRP-XL) in healthy controls (n = 20), patients with Gram-positive (Gram+, n = 15) and Gram-negative (Gram-, n = 17) sepsis. (B) Platelet-monocyte complex (PMC) formation is depicted as the MFI of the platelet marker CD61 on CD14-positive cells. Data depicted are medians with IQR (platelet reactivity) or median with IQR, minimum and maximum values (PMC). * Gram-positive vs. Gram-negative, #Gram-positive vs. healthy controls.
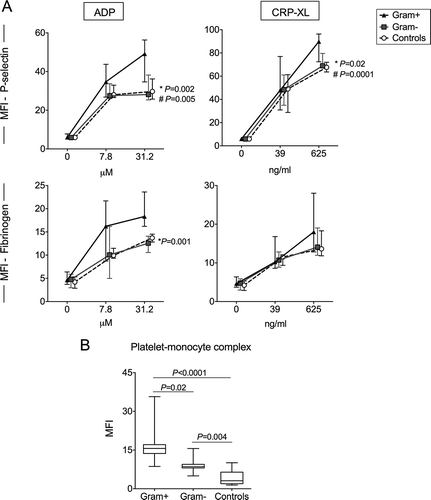
The Gram-positive group had significantly higher PMC formation compared to Gram-negative group. Median (IQR) MFI values of the platelet marker CD61 on CD14-positive cells were 15.6 (13.7–17.1) in the Gram-positive group compared to 8.5 (8.1–9.5; P = 0.02)) in the Gram-negative group and 3.1 (2.0–6.5; P < 0.0001) in controls (). The difference between the Gram-negative group and controls was also significant (P = 0.004). There was a weak positive correlation between the platelet expression of P-selectin and PMC formation in the patients (Spearman, R2 = 0.22; P = 0.008)
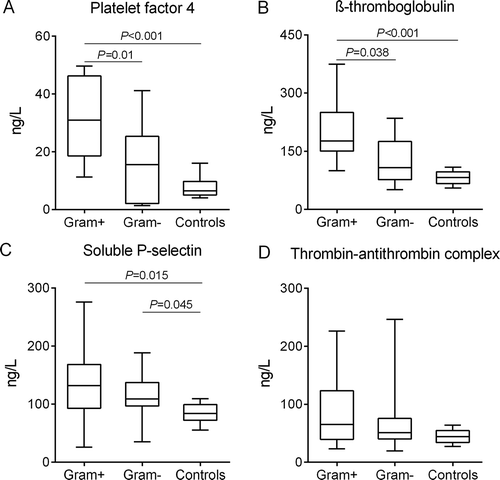
Activated platelets release their α-granule contents, including P-selectin, PF4 and β-TG in the plasma. Plasma concentrations of these proteins were significantly higher in the Gram-positive group compared with the Gram-negative group and the healthy controls. Median (IQR) concentrations of PF4 in these respective groups were 32.1 ng/ml (18.6–46.2 ng/ml), 16.5 ng/ml (2.1–25.3 ng/ml; P = 0.01 vs. Gram-positive) and 7.8 ng/ml (5.0–9.8 ng/ml; P < 0.001 vs. Gram-positive). β-TG concentrations were 200.7 ng/ml (150.6–250 ng/ml), 126.5 ng/ml (77.1–174.9; P = 0.038 vs. Gram-positive) ng/ml and 81.7 ng/ml (66.8–96.8 ng/ml; P = 0.0008 vs. Gram-positive). P-selectin concentrations were 136.4 ng/ml (92.9–168.4 ng/ml), 111.2 ng/ml (96.7–137.1 ng/ml; P = 0.61 vs. Gram-positive) and 84.4 ng/ml (72–99 ng/ml; P = 0.015 vs. Gram-positive). The latter protein was the only soluble platelet parameter that was significantly higher in the Gram-negative group compared to controls (P = 0.045). In contrast, there were no significant differences across the three groups in the plasmatic coagulation marker TAT complexes (). In all patients, soluble P-selectin was weakly correlated with PMC formation (Spearman, R2 = 0.23; P = 0.01). Data of these plasma soluble markers were missing in 2 patients diagnosed with S. aureus sepsis in the Gram-positive group.
Figure 3. Plasma concentrations of platelet and coagulation activation markers. Data depicted are medians with IQR, minimum and maximum values.
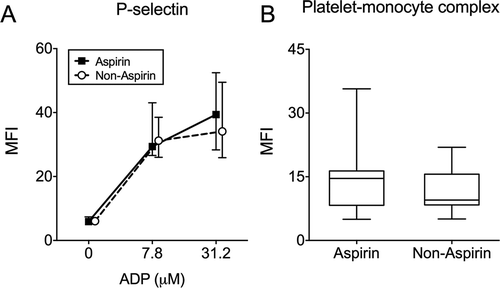
We performed subanalyses within the patient groups and found that those with a S. aureus or Streptococcus spp sepsis in the Gram-positive group had comparable platelet P-selectin expression upon induction with high dose ADP (MFI 47.4, 40.7–53.8 vs. 44.5, 26.9–58.2; P = 0.67) and platelet-fibrinogen binding (MFI 19.0, 14.7–24.0 vs. 17.4, 13.6–22.3; P = 0.61), as well as PMC formation (18.3, 15.3–21.9 vs. 14.4, 12.1–16.5; P = 0.26). Similarly, there were no differences in these parameters between patients with E. coli and other Gram-negative bacteria (data not shown). Aspirin treatment did not influence ADP- and CRP-induced platelet reactivity, PMC formation or the plasma soluble markers (data for ADP-induced platelet reactivity and PMC formation shown in ). There was no significant correlation between ADP-induced P-selectin expression and the time interval between haemostatic tests and the start of antibiotics administration (Spearman, R2 = 0.06, P = 0.07).
Figure 4. Platelet reactivity in aspirin and non-aspirin using patients. Data depicted are medians with IQR, minimum and maximum values.
Discussion
Our data reveal that a sepsis with common Gram-positive pathogens is associated with more pronounced platelet activation, platelet hyperreactivity and PMC formation compared with sepsis due to common Gram-negative pathogens. These differences were observed despite comparable CRP plasma concentrations, suggesting that inflammation by itself is not a major driving force in sepsis-induced platelet activation. In contrast, there were no clear differences in the plasmatic coagulation parameter TAT complexes between the Gram-positive and Gram-negative groups.
The majority of patients in the Gram-positive group had a S. aureus sepsis, whereas E. coli was the most common bacteria in the Gram-negative group. S. aureus and Streptococcus spp. are able to directly bind and activate platelets and different mechanisms through which these pathogens activate platelets have been reviewed recently by Hamzeh-Cognasse et. al. [Citation13]. S. aureus can release toxins or use surface protein such as protein A and clumping factor A, which can subsequently bind platelet receptors directly or indirectly [Citation25,Citation26]. Furthermore, FcγRIIa on platelets is essential for the amplification of their reactivity to these bacteria. The engagement of FcγRIIA by bacteria-bound plasma IgG, together with the integrin αIIbβ3 activation, results in integrin/FcγRIIA ITAM signalling which further triggers ADP, thromboxane A2 (TxA2) and PF4 release and leading to a strong positive feedback cascade of platelet activation [Citation27]. Other S. aureus proteins, including lipoteichoic acid [Citation28], inhibit platelet activation, although our present findings indicate that the platelet-activating potential of S. aureus is dominant in vivo. It was beyond the scope of our current study to characterize the pathways of platelet activation by the isolated pathogens. E. coli was also recently demonstrated to activate platelets in a FcγRIIa-dependent manner [Citation29,Citation30], although ex vivo data of platelet activation by E. coli lipopolysaccharide are conflicting [Citation15–Citation17,Citation31]. Administration of lipopolysaccharide to healthy volunteers also does not appear to strongly activate platelets [Citation32,Citation33], a finding supported by our data of limited platelet activation in patients with E. coli sepsis.
Vascular complications are common in patients with staphylococcal or streptococcal septicaemia [Citation10,Citation34]. We speculate that the strong platelet-activating potential of these micro-organisms contributes to these complications. Platelet function inhibitors might reduce the risk for these complications [Citation35], but this may not be without hazard, as activated platelets are able to limit the growth of and engulf S. aureus [Citation36,Citation37]. In a mouse model, platelet inhibition also reduced killing of S. aureus [Citation38,Citation39].
Limitations of our study include the limited number of patients in both groups and that the haemostatic assays were only performed once after the blood cultures became positive. Gram-negative bacteria can be rapidly killed by antibiotics, and even though no correlation existed between platelet reactivity and the time intervals between haemostatic tests and antibiotic treatment initiation, we cannot exclude that increased platelet activation was present in the Gram-negative group at presentation and that this normalized promptly after administration of antibiotics (i.e. before the blood culture became positive). Furthermore, many Gram-positive and Gram-negative bacteria exist, and our findings cannot be extrapolated to other pathogens that were not included in this study, as their platelet-activating properties may differ. Finally, some patients used aspirin, which inhibits platelet activation via the COX-1 pathway [Citation40]. This was, in our study, not associated with decreased platelet reactivity via the ADP and collagen pathway or PMC formation.
In conclusion, Gram-positive sepsis is associated with marked platelet activation, in contrast to Gram-negative sepsis, and this may contribute to the vascular complications seen in these infections. Given the role of platelets in immunity and host defence, studies are currently undertaken investigating the possible role of platelet inhibition in sepsis patients [Citation41]. It is important that causative pathogens are taken into account, as clear differences exist in the platelet-activating potential of bacteria, which influences the risk of vascular complications.
Conflict of interests
The authors report no conflict of interest.
Acknowledgement
R. Tunjungputri is an awardee of the DIKTI-Neso Fellowship from the Ministry of Research, Technology and Higher Education, Indonesia, and Radboud university medical centre, The Netherlands.
References
- Semple JW, Italiano JE, Jr., Freedman J. Platelets and the immune continuum. Nat Rev Immunol 2011;11:264–274.
- Thomas MR, Storey RF. The role of platelets in inflammation. Thrombosis Haemostasis 2015;114:449–458.
- von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res 2007;100:27–40.
- Ford I, Douglas CW. The role of platelets in infective endocarditis. Platelets 1997;8:285–294.
- Smeeth L, Thomas SL, Hall AJ, Hubbard R, Farrington P, Vallance P. Risk of myocardial infarction and stroke after acute infection or vaccination. N Eng J Med 2004;351:2611–2618.
- Meier CR, Jick SS, Derby LE, Vasilakis C, Jick H. Acute respiratory-tract infections and risk of first-time acute myocardial infarction. Lancet 1998;351:1467–1471.
- Clayton TC, Thompson M, Meade TW. Recent respiratory infection and risk of cardiovascular disease: case-control study through a general practice database. Eur Heart J 2008;29:96–103.
- Weidenmaier C, Peschel A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat Rev Microbiol 2008;6:276–287.
- Musher DM, Rueda AM, Kaka AS, Mapara SM. The association between pneumococcal pneumonia and acute cardiac events. Clin Infect Dis 2007;45:158–165.
- Dalager-Pedersen M, Sogaard M, Schonheyder HC, Nielsen H, Thomsen RW. Risk for myocardial infarction and stroke after community-acquired bacteremia: a 20-year population-based cohort study. Circulation 2014;129:1387–1396.
- Dong M, Liu T, Li G. Association between acute infections and risk of acute coronary syndrome: a meta-analysis. Int J Cardiol 2011;147:479–482.
- Murdoch DR, Corey GR, Hoen B, Miro JM, Fowler VG, Jr., Bayer AS, et al. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the International Collaboration on Endocarditis-Prospective Cohort Study. Arch Internal Med 2009;169:463–473.
- Hamzeh-Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, Garraud O. Platelets and Infections–Complex Interactions with Bacteria. Front Immun 2015;6:82.
- Tunjungputri RN, de Jonge MI, de Greeff A, van Selm S, Buys H, Harders-Westerveen JF, et al. Invasive pneumococcal disease leads to activation and hyperreactivity of platelets. Thrombosis Res 2016;144:123–126.
- Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MK, et al. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thrombosis Haemostasis 2005;94:831–838.
- Sheu JR, Hung WC, Kan YC, Lee YM, Yen MH. Mechanisms involved in the antiplatelet activity of Escherichia coli lipopolysaccharide in human platelets. Br J Haematol 1998;103:29–38.
- Montrucchio G, Bosco O, Del Sorbo L, Pecetto PF, Lupia E, Goffi A, et al. Mechanisms of the priming effect of low doses of lipopoly-saccharides on leukocyte-dependent platelet aggregation in whole blood. Thrombosis Haemostasis 2003;90:872–881.
- Damien P, Cognasse F, Eyraud MA, Arthaud CA, Pozzetto B, Garraud O, et al. LPS stimulation of purified human platelets is partly dependent on plasma soluble CD14 to secrete their main secreted product, soluble-CD40-Ligand. BMC Immun 2015;16:3.
- Klinkhardt U, Bauersachs R, Adams J, Graff J, Lindhoff-Last E, Harder S. Clopidogrel but not aspirin reduces P-selectin expression and formation of platelet-leukocyte aggregates in patients with atherosclerotic vascular disease. Clin Pharmacol Ther 2003;73:232–241.
- Storey RF, Judge HM, Wilcox RG, Heptinstall S. Inhibition of ADP-induced P-selectin expression and platelet-leukocyte conjugate formation by clopidogrel and the P2Y12 receptor antagonist AR-C69931MX but not aspirin. Thrombosis Haemostasis 2002;88:488–494.
- Tunjungputri RN, Van Der Ven AJ, Schonsberg A, Mathan TS, Koopmans P, Roest M, et al. Reduced platelet hyperreactivity and platelet-monocyte aggregation in HIV-infected individuals receiving a raltegravir-based regimen. AIDS (London, England). 2014;28:2091–2096.
- van Bladel ER, Laarhoven AG, van der Heijden LB, Heitink-Pollé KM, Porcelijn L, van der Schoot CE, et al. Functional platelet defects in children with severe chronic ITP as tested with two novel assays applicable for low platelet counts. Blood. 2014: blood-2013-08-519686.
- Michelson AD, Barnard MR, Krueger LA, Valeri CR, Furman MI. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 2001;104:1533–1537.
- Snoep JD, Roest M, Barendrecht AD, De Groot PG, Rosendaal FR, Van Der Bom JG. High platelet reactivity is associated with myocardial infarction in premenopausal women: a population-based case-control study. JTH 2010;8:906–913.
- Kerrigan SW, Kaw G, Hogan M, Penadés J, Litt D, Fitzgerald DJ, et al. Multiple mechanisms for the activation of human platelet aggregation by Staphylococcus aureus: roles for the clumping factors ClfA and ClfB, the serine–aspartate repeat protein SdrE and protein A. Mol Microbiol 2002;44:1033–1044.
- Kerrigan SW, Clarke N, Loughman A, Meade G, Foster TJ, Cox D. Molecular Basis for Staphylococcus aureus–Mediated Platelet Aggregate Formation Under Arterial Shear In Vitro. Arteriosclerosis, Thrombosis, Vasc Biol 2008;28:335–340.
- Arman M, Krauel K, Tilley DO, Weber C, Cox D, Greinacher A, et al. Amplification of bacteria-induced platelet activation is triggered by FcgammaRIIA, integrin alphaIIbbeta3, and platelet factor 4. Blood 2014;123:3166–3174.
- Sheu JR, Lee CR, Lin CH, Hsiao G, Ko WC, Chen YC, et al. Mechanisms involved in the antiplatelet activity of Staphylococcus aureus lipoteichoic acid in human platelets. Thrombosis Haemostasis 2000;83:777–784.
- Moriarty RD, Cox A, McCall M, Smith SG, Cox D. Escherichia coli induces platelet aggregation in an FcgammaRIIa-dependent manner. JTH 2016;14:797–806.
- Watson CN, Kerrigan SW, Cox D, Henderson IR, Watson SP, Arman M. Human platelet activation by Escherichia coli: roles for FcgammaRIIA and integrin alphaIIbbeta3. Platelets 2016; 27:535–540.
- Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, et al. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol (Baltimore, Md: 1950). 2009;182:7997–8004.
- Schrottmaier WC, Kral JB, Zeitlinger M, Salzmann M, Jilma B, Assinger A. Platelet activation at the onset of human endotoxemia is undetectable in vivo. Platelets 2016; 27:479–483.
- Thomas MR, Outteridge SN, Ajjan RA, Phoenix F, Sangha GK, Faulkner RE, et al. Platelet P2Y12 Inhibitors Reduce Systemic Inflammation and Its Prothrombotic Effects in an Experimental Human Model. Arteriosclerosis, Thrombosis, Vasc Biol 2015;35:2562–2570.
- Corrales-Medina VF, Fatemi O, Serpa J, Valayam J, Bozkurt B, Madjid M, et al. The association between Staphylococcus aureus bacteremia and acute myocardial infarction. Scand J Infect Dis 2009;41:511–514.
- Anavekar NS, Tleyjeh IM, Anavekar NS, Mirzoyev Z, Steckelberg JM, Haddad C, et al. Impact of prior antiplatelet therapy on risk of embolism in infective endocarditis. Clin Infect Dis 2007;44:1180–1186.
- Youssefian T, Drouin A, Masse JM, Guichard J, Cramer EM. Host defense role of platelets: engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood 2002;99:4021–4029.
- Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, et al. Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog 2011;7:e1002355.
- Wuescher LM, Takashima A, Worth RG. A novel conditional platelet depletion mouse model reveals the importance of platelets in protection against Staphylococcus aureus bacteremia. JTH 2015;13:303–313.
- Zhang X, Liu Y, Gao Y, Dong J, Mu C, Lu Q, et al. Inhibiting platelets aggregation could aggravate the acute infection caused by Staphylococcus aureus. Platelets 2011;22:228–236.
- Schror K. Aspirin and platelets: the antiplatelet action of aspirin and its role in thrombosis treatment and prophylaxis. Semin Thrombosis Hemostasis 1997;23:349–356.
- Thomas MR, Storey RF. Effect of P2Y12 inhibitors on inflammation and immunity. Thrombosis Haemostasis 2015;114:490–497.