
Abstract
Involvement of signal transducer and activator of transcription 3 (STAT3) in inflammation is well known. Recently, a role for STAT3 in platelet activation and platelet production has been suggested. Platelets exhibit important immune functions and engagement of STAT3 in platelet physiology may link inflammation and hemostasis. This study investigated the effects of STAT3 loss-of-function mutations and single nucleotide polymorphisms (SNPs) in STAT3 on glycoprotein VI (GPVI)-mediated platelet activation and platelet numbers in humans. Two cohorts were studied. The first cohort concerned patients with STAT3 loss-of-function mutations. Platelet numbers were investigated in eight patients and GPVI-mediated platelet activation was functionally tested in four patients. Additional experiments were performed to investigate underlying mechanisms. The second cohort concerned 334 healthy volunteers and investigated the consequences of SNPs in STAT3 on GPVI-mediated platelet activation and platelet numbers. Platelet activation was lower in STAT3 loss-of-function patients at baseline and after stimulation of the GPVI receptor, reflected by decreased P-selectin expression. This was independent of gene transcription. Blockade of the adenosine di-phosphate (ADP) pathway resulted in a further decrease of P-selectin expression, particularly in STAT3 loss-of-function patients. In contrast, the SNPs in STAT3 did not influence GPVI-mediated platelet activation. Also, platelet numbers were not affected by STAT3 loss-of-function mutations, nor was there an association with the SNPs. In conclusion, STAT3 signaling does not seem to play a major role in thrombopoiesis. We confirm that STAT3 is involved in GPVI-mediated platelet activation in humans, independent of gene transcription. GPVI-mediated platelet activation is highly dependent on secondary ADP release. Our findings suggest that STAT3 modulation may affect inflammation, hemostasis, and their interaction.
Introduction
28 September 2017 STAT3 was shown to play a role in platelet activation [Citation9–Citation11] and in thrombopoiesis [Citation12]. Zhou and colleagues reported that platelet activation via surface receptor glycoprotein VI (GPVI) is impaired in platelet STAT3-knockout mice [Citation9]. Additionally, ex vivo STAT3 inhibition by pharmacological inhibitors resulted in decreased platelet activation and less thrombus formation after stimulation of GPVI with platelet agonist collagen-related peptide (CRP) in humans [Citation9]. Grozovsky and colleagues showed involvement of STAT3 in platelet production [Citation12]. Binding of desialylated platelets to the Ashwell–Morell receptor induced hepatic thrombopoietin (TPO) via a mechanism dependent on STAT3 in mice [Citation12]. These studies suggest that STAT3 may be involved in primordial platelet physiology; however, most evidence is derived from mouse models and cell lines: further validation of its importance in humans is therefore warranted.
This study investigated involvement of STAT3 in platelet activation and platelet numbers in humans. Two independent cohorts were studied. The first cohort concerned patients with STAT3 loss-of-function mutations. STAT3 loss of function is rare but nonetheless observed in humans and results in the hyper IgE syndrome, an immune deficiency disorder associated with recurrent infections, eczema, mucocutaneous candidiasis, and extreme elevations of serum IgE [Citation13,Citation14]. Patients with STAT3 loss of function are deficient in STAT3-dependent cytokines, such as interleukin (IL)-17. It is unknown whether thrombopoiesis and platelet function are affected in humans with mutations in STAT3. The second cohort concerned healthy human volunteers and investigated the effects of single nucleotide polymorphisms (SNPs) in STAT3 on platelet activation and platelet numbers. These data are important to understand its function on a population level. We demonstrate that both mutations and SNPs in STAT3 do not influence platelet numbers in humans, whereas STAT3 loss-of-function mutations do affect GPVI-mediated platelet activation by CRP. Additionally, we observed that GPVI-mediated platelet activation was largely dependent on secondary stimulation by adenosine di-phosphate (ADP), thereby partially restoring the defect observed in STAT3 loss-of-function patients.
Methods
Patients selection
Cohort 1. Subjects with STAT3 loss-of-function mutations
Autosomal-dominant cases of hyper IgE syndrome (AD-HIES) are caused by specific mutations that result in nonfunctional STAT3 activity [Citation15]. Most mutations are found at positions that are well known for STAT3 function, such as the Src homology (SH2) domain that enables recruitment and binding of STAT3 to the activated receptor and the DNA-binding domain that enables binding of STAT3 to DNA target sites [Citation14]. The vast majority of AD-HIES cases are due to these heterozygous mutations resulting in absent STAT3 activity; however, several other mutations have been described [Citation13,Citation16]. To examine the effects of STAT3 loss-of-function mutations on platelet numbers, medical records of eight confirmed heterozygous AD-HIES patients were consulted. Additionally, we were able to functionally assess GPVI-mediated platelet activation in 4 of these patients and in 10 control subjects. IL-17 production has been previously tested in peripheral blood mononuclear cells of these patients and the IL-17 deficiency served as a further confirmation of the STAT3 loss of function [Citation17]. Control subjects were healthy individuals who were age and gender matched with AD-HIES patients. No genotyping for specific STAT3 mutations was performed in control subjects because the incidence is exceptionally low and severe medical complaints manifest early in life. The control subjects did not experience any medical complaints during recruitment. Subjects were recruited under a protocol approved by the Institutional Review Board of Radboud University Nijmegen Medical Center. AD-HIES patients are referred to as STAT3 loss-of-function patients.
Cohort 2. STAT3-related SNPs in healthy subjects
Several SNPs in STAT3 that are associated with inflammatory diseases were identified from literature [Citation18–Citation20]. In contrast to the STAT3 loss-of-function mutations, the SNPs in STAT3 are associated with inflammatory diseases that are accompanied by increased IL-17 levels. Differentiation of Th-17 cells is dependent on STAT3, which suggests increased STAT3 activity [Citation20]. We determined these SNPs using DNA from the 500 functional genomics cohort and correlated these with GPVI-mediated platelet activation and platelet numbers. This cohort consists of 534 healthy individuals of Caucasian origin and is part of the Human Functional Genomics Project aimed at characterizing variations in immune function [Citation21,Citation22]. More detailed information on the study design and the characteristics of study participants can be found in previous publications [Citation22,Citation23]. Individuals were recruited under a protocol approved by the Institutional Review Board of Radboud University Nijmegen Medical Center.
Experiments cohort 1
Platelet activation and responsiveness assay
Blood from STAT3 loss-of-function patients during stable disease and healthy subjects was collected in 3.2% sodium citrate vacutainer tubes (Becton Dickinson, Franklin Lakes, New Jersey, USA). Whole blood was centrifugated for 15 min at 156g without break to obtain platelet-rich plasma (PRP). Platelet concentration was adjusted to 300 × 109/l by addition of autologous platelet-poor plasma (PPP). PPP was obtained after centrifugation of whole blood at 3800 RPM for 10 min. PRP was rested for 1 h before platelet functions were assessed.
Platelet expression of activation marker P-selectin was measured in PRP at baseline and after incubation for 20 min with different concentrations of platelet agonist cross-linked CRP (kind gift from Prof. Dr. R. Farndale, Cambridge, UK) to stimulate the GPVI surface receptor. Thrombin receptor-activating peptide-6 (TRAP) (Sigma-Aldrich, Saint Louis, Missouri, USA), an agonist for the thrombin receptor proteinase-activated receptor-1, served as a positive control. Samples were incubated with nine concentrations of CRP (range 1.3–320 ng/ml) and two concentrations of TRAP (9.78 and 156 µm) together with antibodies for flow cytometry. The reaction was stopped by addition of 0.2% paraformaldehyde. P-selectin expression was measured with a Becton Dickinson flow cytometer. In order to identify platelets, platelets were gated based on forward and sideward scatter and additional gating was performed based on expression of platelet surface marker CD61 (PC7 labeled anti-CD61, Beckman Coulter, Brea, California, USA). To determine platelet activation, the mean fluorescence intensity (MFI) exceeding that of the matched isotype of P-selectin (PE labeled anti-CD62P, Bio-legend, San Diego, California, USA) was determined.
Inhibition of ADP pathway with apyrase
Activation via the GPVI surface receptor results in platelet degranulation of dense granules thereby releasing biologically active substances, most notably ADP. ADP can activate platelets via a secondary pathway. In order to avoid skewing of the responses by platelet activation via this positive feedback loop, additional experiments were performed after preincubation with apyrase, to block the ADP pathway. Apyrase was purchased from Sigma-Aldrich (Saint Louis, Missouri, USA). For experiments, a final concentration 5 U/ml was used. After incubation with apyrase, PRP samples were stimulated with CRP at concentrations 320, 80, and 20 ng/ml in combination with the antibodies for flow cytometry. The same protocol and gating strategies as described above were used.
Inhibition of transcription during platelet activation
STAT3, as a transcription factor, regulates DNA transcription and exerts its effects after phosphorylation, dimerization, and translocation to the nucleus [Citation24]. Platelets do not have a nucleus and exclusively contain mitochondrial DNA. Experiments with transcription blocker actinomycin were performed to investigate the role of DNA transcription in this process. Actinomycin was purchased from Sigma-Aldrich (Saint Louis, Missouri, USA) and dissolved in DMSO, stock concentration 6270 µg/ml. PRP was preincubated with actinomycin D in two concentrations (50 and 5 µg) and DMSO control, before samples were stimulated with CRP. The CRP concentration of 40 ng/ml was chosen as it was estimated that platelet responses would be in the steep part of the platelet reactivity curve. The same protocol for stimulation and similar gating strategies as described above was used for measurement of P-selectin expression.
Platelet content and plasma markers
Soluble plasma markers and platelet content of P-selectin, fibrinogen were determined by ELISA. For platelet content, PRP (concentration 300 × 109 platelets/ml) was freeze thawed for three cycles to fragment the platelets. Samples were centrifugated at room temperature for 5 min at 5000 RPM in an ultracentrifuge to spin down large particles. Supernatants were used for ELISA. PPP was used for plasma markers. Soluble P-selectin and soluble fibrinogen were measured using a human P-selectin/CD62P duoset ELISA kit (R&D systems, Europe, Abingdon, UK) and a human fibrinogen ELISA kit (Abcam, Europe, Cambridge, UK), respectively. TPO was measured with a human TPO Quantikine ELISA kit (R&D systems, Europe, Abingdon, UK).
Experiments cohort 2
SNPs in STAT3
Genotyping of the SNPs was performed using a commercially available SNP chip, Illumina HumanOmniExpressExome-8 v.1.0, methods previously reported by Li et al. [Citation23]. In short, genotype calling was performed using Optical 0.7.0 [Citation25]. Call rates less than ≤0.99 were excluded from the dataset, as were samples with a Hardy–Weinburg equilibrium ≤0.0001, call rate ≤0.99, and minor allele frequencies (MAF) ≤0.001. In total, 483 samples were included for further imputation, as described previously. Data were extracted for the following SNPs: rs744166, rs3816769, and rs4796793. Presence of the SNP was related to platelet activation in response to CRP and to platelet numbers.
Platelet activation and responsiveness assay
For assessment of platelet activation and responsiveness, platelet P-selectin expression was measured in whole blood at baseline and after incubation for 20 min with seven different concentrations of platelet agonists CRP, together with an antibody mix for flow cytometry. The same protocol and gating strategies as described above were used for measurement of P-selectin expression. The area under the curve (AUC) of the P-selectin expression after stimulation (MFI) was correlated with the SNP data. In total, data were available for 334 healthy human volunteers.
Statistical analysis
Cohort 1. Quantitative data are expressed as median with interquartile range (IQR) and were analyzed with GraphPad Prism 5. Repeated measures like platelet responsiveness upon stimulation were analyzed using a two-way ANOVA in order to correct for the multiple independent observations. To compare singular measurements between groups Mann–Whitney U tests were performed for non-normally distributed data.
Cohort 2. Platelet numbers were normally distributed, in contrast to P-selectin expression. To normalize P-selectin expression (AUC), data were log transformed. Formal correction for age and gender effects in both outcome parameters was performed. SNP genotypes were coded 0, 1, and 2, before linear regression analyses were performed with SPSS with use of dummy variables.
p Values < 0.05 were considered statistically significant.
Results
Characteristics of study participants
Main characteristics of the two study cohorts are presented in . The first cohort concerned eight STAT3 loss-of-function patients, five patients had a mutation in the DNA-binding domain, two in the SH2-domain, and one patient had a mutation in the Linker domain of STAT3. The second cohort involved 483 healthy human volunteers of which 334 participants data on SNPs and GPVI-mediated platelet activation were available. A more detailed description of the specific mutations in the STAT3 loss-of-function patients can be found in .
Table I. Patient characteristics.
Table II. STAT3 loss-of-function mutations.
Less platelet activation and GPVI-mediated platelet responsiveness in STAT3 loss-of-function patients
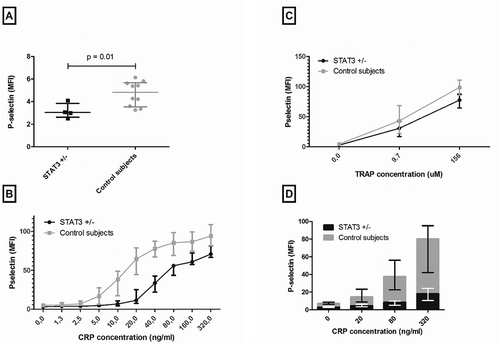
To investigate the involvement of STAT3 in platelet activation, we determined baseline platelet activation in STAT3 loss-of-function patients. Platelet activation, determined by P-selectin expression, was significantly lower in STAT3 loss-of-function patients compared to control subjects (median MFI 3.03 [IQR 2.62–3.84] vs. 4.82 [IQR 3.53–5.67], p = 0.01) (). Next, we investigated whether collagen-induced platelet activation by stimulation of GPVI surface receptor with CRP was affected. Stimulation of the GPVI receptor with different concentrations of CRP (range 1.3–320 ng/ml) resulted in significantly lower P-selectin expression in STAT3 loss-of-function patients compared to control subjects (p < 0.001) (). To examine whether this observation was due to an intrinsic platelet defect or whether it resulted from the theoretical possibility of defective production of factors that prime platelets, platelets were stimulated with another agonist, TRAP, as a control. Although platelet responses were slightly lower with TRAP (), this effect was not statistically significant after correction for lower baseline P-selectin expression, suggesting a largely intrinsic defect in platelets.
Figure 1. Functional assessment of GPVI-mediated platelet activation in STAT3 loss-of-function patients.
(A) Platelet P-selectin expression in unstimulated samples (median MFI 3.03 (IQR 2.62–3.84) in STAT3 loss-of-function versus 4.82 (IQR 3.53–5.67) in control subjects, p = 0.01). (B) Platelet P-selectin expression in GPVI-mediated platelet activation by CRP stimulation (p < 0.001). (C) Platelet P-selectin expression in PAR-1-mediated platelet activation by TRAP (p = 0.054). (D) P-selectin expression in GPVI-mediated platelet activation by CRP after blockade of the ADP pathway by apyrase. Data are presented as median with IQR (ADP: adenosine 5′ diphosphate; CRP: collagen-related peptide; GPVI: glycoprotein VI; MFI: mean fluorescence intensity; TRAP: thrombin receptor-activating peptide-6).
Inhibition of the ADP pathway further attenuates platelet responsiveness in STAT3 loss-of-function patients
GPVI-mediated platelet activation results in platelet degranulation, thereby releasing biologically active substances, in particular ADP from dense granules [Citation26]. In order to investigate dependency of the GPVI-mediated platelet activation pathway on secondary ADP release, we incubated samples with apyrase to block this pathway. Stimulation of the GPVI receptor by different concentrations of CRP after incubation of samples with apyrase resulted in considerably lower P-selectin expression in both groups. Inhibition of the ADP pathway seems to have a more profound effect in STAT3 loss-of-function patients than in healthy subjects, as the relative increase from baseline to the highest CRP concentration was 5.7-fold in STAT3 loss-of-function patients compared to 11.4-fold in control subjects ().
STAT3 signaling in GPVI-mediated platelet activation is independent of gene transcription
Platelets do not have a DNA-containing nucleus; however, platelets do possess functional mitochondrial DNA [Citation26]. In order to exclude the theoretical possibility of involvement of novel DNA transcription in platelet activation in response to CRP stimulation, transcription was inhibited by general transcription blocker actinomycin D. After incubation of PRP samples with actinomycin D, samples were stimulated with CRP. No changes in expression of P-selectin were seen after inhibition of transcription in both patients and control subjects (data not shown).
No differences in platelet content and soluble markers were found in STAT3 loss-of-function patients
Previous experiments suggest an intrinsic platelet defect in patients with STAT3 loss-of-function mutations. In addition, inhibition of the ADP pathway had a relatively higher impact on GPVI-mediated platelet activation in STAT3 loss-of-function patients. To examine whether these observations were a result of alterations in granule content, we measured P-selectin and fibrinogen in platelet lysates. No differences were seen in platelet content for P-selectin and fibrinogen (327.0 ng/ml [IQR 153.9–523.5] in patients vs. 261.1 ng/ml [IQR 204.3–325.5] in control subjects, p-value 0.84 and 2420 µg/ml [IQR 1786–3813] in patients, and 2092 µg/ml [IQR 1038–2938] in control subjects, p-value 0.52, respectively). This suggests that previous observations are more likely due to lower platelet granule release in response to stimulation than alterations in granule content.
Lastly, P-selectin and fibrinogen in plasma was determined as a measure for platelet granules release. Interestingly, no significant differences were found in soluble plasma P-selectin and fibrinogen between patients and control subjects (median 41.7 ng/ml [IQR 27.0–47.3] vs. 27.9 ng/ml [21.9–44.2], p-value 0.45 and mean 2513 µg/ml [IQR 1826–3016] vs. 2640 µg/ml [IQR 2240–3114], p-value 0.74, respectively).
SNPs in STAT3 do not correlate with GPVI-mediated platelet activation in healthy volunteers
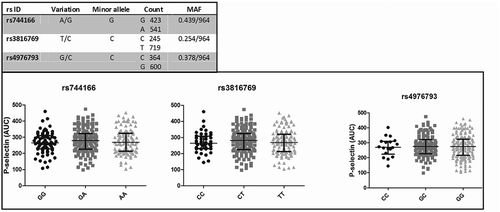
To investigate the involvement of STAT3 on platelet activation on a population level, several SNPs in STAT3 that are associated with inflammatory diseases were investigated. The three SNPs, rs744166, rs3816769, and rs4796793, were determined in the second cohort, concerning healthy volunteers. These SNPs are known from literature to be functional and are likely to influence STAT3 activity in relation to several diseases, such as Crohn’s disease, ulcerative colitis, and autoimmune thyroid disease [Citation18–Citation20]. The MAF of rs744166, rs3816769, and rs4796793 were C = 0.439/964, C = 0.254/964, and C = 0.378/964, respectively. The MAFs were similar to the MAFs reported in the NCBI database (http://www.ncbi.nlm.nih.gov/SNP), except for rs3816769 with a MAF of C = 382/1912 in the NCBI database.
Linear regression analysis was performed for SNP genotype and platelet activation in response to GPVI stimulation (AUC). No significant correlations could be detected for rs744166, rs3816769, and rs4796793 with GPVI-mediated platelet activation (p > 0.05). The SNP characteristics and the relation between the SNPs and GPVI-mediated platelet activation are shown in . In contrast to previous observations, SNPs in STAT3 do not seem to affect GPVI-mediated platelet activation.
Figure 2. SNPs rs744166, rs3816769, and rs4796793 and P-selectin expression (AUC).
SNP characteristics are shown in the upper figure. P-selectin expression (AUC of MFI) after GPVI-mediated platelet activation by seven concentrations of CRP is shown in the lower figure. Data are presented as median with IQR (AUC: Area under the curve; CRP: collagen-related peptide, GPVI: glycoprotein VI; IQR: interquartile range; MAF: minor allele frequency; MFI: mean fluorescence intensity; SNP: single nucleotide polymorphism).
Platelet numbers are not influenced by STAT3 mutations and STAT3 SNPs
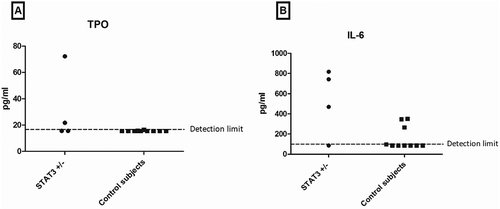
Lastly, we studied platelet production. Platelet production is primarily regulated by TPO concentrations in blood and it has recently been suggested that hepatic TPO production is dependent on signaling via STAT3 [Citation12]. Platelet numbers retrieved from medical records of eight confirmed STAT3 loss-of-function patients were mostly within the normal range (). Interestingly, thrombocytosis was observed in two patients during an acute infection. Thrombocytopenia was not observed in STAT3 loss-of-function patients. In addition, plasma TPO levels were determined in the 4 functionally assessed STAT3 loss-of-function patients and the 10 healthy subjects. Although most values were below the detection limit of 15.6 pg/ml, two STAT3 loss-of-function patients had detectable TPO levels (24.0 and 79.7 pg/ml) (). Several studies suggest that IL-6 stimulates thrombopoiesis [Citation27,Citation28]. Therefore, we determined IL-6 levels in plasma. A trend for higher IL-6 was observed in STAT3 loss-of-function patients ().
Table III. Median platelet numbers (range).
Figure 3. TPO and IL-6 levels in plasma.
(A) Plasma TPO levels in STAT3 loss-of-function patients and control subjects. (B) Plasma IL-6 expression in STAT3 loss-of-function patients and control subjects (IL-6: Interleukin-6; TPO: thrombopoietin).
Also the effects of the SNPs in STAT3 were examined. Consistently with the observation in the STAT3 loss-of-function patients, no significant influence of the SNPs on platelet numbers was observed in 334 healthy subjects (). Importantly, both thrombocytopenia and thrombocytosis were rarely observed.
Discussion
The present study investigated the involvement of STAT3 in GPVI-mediated platelet activation and platelet numbers in humans. Our findings indicate that GPVI-mediated platelet activation is affected by STAT3 loss-of-function mutations confirming its involvement in platelet function in humans. In contrast, neither clinically relevant STAT3 loss-of-function mutations nor SNPs in STAT3 affected platelet numbers in humans.
Further experiments were performed to increase our understanding of its function. GPVI-mediated platelet activation was shown to be largely dependent on secondary stimulation by platelet ADP release, both in patients and in healthy subjects. Blockade of this mechanism strongly attenuated platelet responses, particularly in STAT3 loss-of-function patients. These observations were independent of gene transcription, as previously reported [Citation9]. Although STAT3 is commonly known as a transcription factor, its involvement in platelet activation appears to be non-transcriptional and few non-transcriptional properties of STAT3 have been previously described [Citation29,Citation30]. SNPs in STAT3 did not directly affect GPVI-mediated platelet activation in a healthy population. In some inflammatory diseases, the effects of SNPs in STAT3 on platelet activation may become apparent; however, in healthy individuals, robust mechanisms seem to balance inflammation and hemostasis.
Interestingly, P-selectin levels in plasma were not different between STAT3 loss-of-function patients and control subjects, whereas platelet degranulation was decreased in patients. Another source for soluble P-selectin is the vascular endothelium [Citation31]. Both vascular abnormalities and endothelial dysfunction are common in hyper IgE syndrome and may explain this observation [Citation32].
With the lack of an effect on platelet numbers, we report an opposite effect compared to a previous study that suggests an important role for STAT3 in the hepatocytic production of TPO, the most important regulator of platelet formation by megakaryocytes [Citation12]. In addition to a mouse model, it was shown that desialylated platelets are taken up by a human HepG2 cells and these cells subsequently produce TPO in a Janus kinase-2 (JAK2)/STAT3-dependent manner [Citation12]. In contrast, we did not find a decrease in platelet numbers in patients with defective STAT3 signaling; moreover, thrombocytosis was observed in STAT3 loss-of-function patients during acute infections. TPO levels were detectable in plasma in two patients, whereas these were low or undetectable in control subjects. A direct comparison between groups is limited by the fact that TPO was undetectable in most subjects and these results should be interpreted with caution. A trend was seen for increased IL-6 in patients and this may also stimulate thrombopoiesis [Citation27,Citation28]. Our findings suggest that in vivo other mechanisms also regulate platelet production in humans.
The observation that STAT3 is involved in GPVI-mediated platelet activation further validates that the mechanism reported by Zhou and colleagues may be functional and relevant in humans too [Citation9]. Involvement of STAT3 in platelet activation may provide a relevant link between inflammation and hemostasis. Increased activation of the STAT3 signaling pathway has been described in cancer and many inflammatory diseases, including cardiovascular diseases [Citation4–Citation8,Citation33]. Cardiovascular disease is the leading cause of death worldwide and interestingly, risk for cardiovascular diseases is strongly increased in many inflammatory diseases [Citation34]. To examine whether GPVI-mediated platelet activation was affected by SNPs in STAT3, we studied a cohort of healthy human volunteers; however, no relation was observed. The role of STAT3 activation in inflammatory diseases and its consequences for platelet activation needs further exploration. Especially since it is unknown if the SNPs in STAT3 affect STAT3 function in healthy subjects, or whether aberrant STAT3 function becomes apparent in inflammatory diseases.
Blockade of STAT3 is currently being explored as a new therapy for cancer [Citation35,Citation36]. Several STAT3 inhibitors have recently proceeded into clinical trials and also drugs that block JAK2, such as Ruxolitinib [Citation35,Citation37,Citation38]. It is important that involvement of STAT3 in platelet activation is recognized. Bleeding complications may occur, especially in patients who are concurrently treated with platelet inhibitors that target other pathways such as Ticagrelor and Clopidogrel, targets of the ADP receptor, P2Y12. GPVI-mediated platelet activation is highly dependent on secondary activation via ADP and suppression of both pathways may have important consequences in terms of bleeding [Citation39–Citation41].
The relevance of the functional impairment in patients with AD-HIES seems to be limited due to the positive feedback loop by ADP, which partially restores platelet responses. Although severe bleeding complications are reported, they mostly coincide with opportunistic infections in AD-HIES patients who often have bronchiectasis, both features known to provoke pulmonary hemorrhages [Citation42,Citation43]. Interestingly, in one AD-HIES patient, a spontaneous bleeding was recorded that could not be explained from a clinical perspective. The bleeding occurred in the iliopsoas muscle and was not preceded by any kind of trauma. Examination of the medical history of the other patients did not reveal any unexplained spontaneous bleedings.
Our study has a few limitations. The first limitation is that we were unable to relate platelet responses directly to STAT3 function and different STAT3 isoforms. STAT3 has several isoforms that mostly result from alternative mRNA splicing. These isoforms can have distinct functions; STAT3β, for example, lacks the Ser727 phosporylation site and is thought to be a negative regulator [Citation44]. The exact mechanisms by which STAT3 mutations cause STAT3 loss-of-function are still largely unknown [Citation14]. It would have been of great interest to directly relate STAT3 isoforms and STAT3 activity to GPVI-mediated platelet responses. Also the question whether there is a compensatory increase in other STATs needs further investigation, as STATs can both form homodimers and heterodimers with different functions [Citation44]. The second limitation is the limited sample size of STAT3 loss-of-function patients (AD-HIES patients). This makes our findings less robust. AD-HIES is a very rare disease with an estimated prevalence of 1:100 000 [Citation14,Citation45]. It has to be noted that the effects observed were consistent in patients and in different concentrations of CRP. Lastly, STAT3 loss-of-function patients are treated with prophylactic antimicrobial therapy and we were unable to correct for those differences in the healthy subjects. Therefore, we cannot completely exclude a potential effect of medication on our findings.
In conclusion, our findings indicate that STAT3 is involved in GPVI-mediated platelet activation by CRP in humans. Additionally, GPVI-mediated platelet activation was shown to be largely dependent on secondary stimulation by ADP. In contrast, neither clinically relevant STAT3 loss-of-function mutations nor SNPs affecting STAT3 activity influence platelet numbers in humans. These data improve our understanding on the interaction between inflammation and hemostasis and suggest that STAT3 modulation may affect both inflammation and hemostasis and their interaction in humans.
Declaration of Interest
Authors declare no competing interests.
Acknowledgments
We would like to thank all patients and healthy volunteers that participated in this trial.
References
- Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol 2015;194:21–27.
- Vogel TP, Milner JD, Cooper MA. The Ying and Yang of STAT3 in human disease. J Clin Immunol 2015;35:615–623.
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol 2009;27:485–517.
- Lu D, Liu L, Ji X, Gao Y, Chen X, Liu Y, Liu Y, Zhao X, Li Y, Li Y, et al. The phosphatase DUSP2 controls the activity of the transcription activator STAT3 and regulates TH17 differentiation. Nat Immunol 2015;16:1263–1273.
- Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: a review. Int J Cancer 2016;138:2570–2578.
- Peyser ND, Freilino M, Wang L, Zeng Y, Li H, Johnson DE, Grandis JR. Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene 2016;35:1163–1169.
- Haapaniemi EM, Kaustio M, Rajala HL, Van Adrichem AJ, Kainulainen L, Glumoff V, Doffinger R, Kuusanmaki H, Heiskanen-Kosma T, Trotta L, et al. Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood 2015;125:639–648.
- Gao W, McCormick J, Connolly M, Balogh E, Veale DJ, Fearon U. Hypoxia and STAT3 signalling interactions regulate pro-inflammatory pathways in rheumatoid arthritis. Ann Rheum Dis 2015;74:1275–1283.
- Zhou Z, Gushiken FC, Bolgiano D, Salsbery BJ, Aghakasiri N, Jing N, Wu X, Vijayan KV, Rumbaut RE, Adachi R, et al. Signal transducer and activator of transcription 3 (STAT3) regulates collagen-induced platelet aggregation independently of its transcription factor activity. Circulation 2013;127:476–485.
- Yuan H, Houck KL, Tian Y, Bharadwaj U, Hull K, Zhou Z, Zhu M, Wu X, Tweardy DJ, Romo D, et al. Piperlongumine blocks JAK2-STAT3 to inhibit collagen-induced platelet reactivity independent of reactive oxygen species. PLoS One 2015;10:e0143964.
- Lu WJ, Lin KC, Huang SY, Thomas PA, Wu YH, Wu HC, Lin KH, Sheu JR. Role of a Janus kinase 2-dependent signaling pathway in platelet activation. Thromb Res 2014;133:1088–1096.
- Grozovsky R, Begonja AJ, Liu K, Visner G, Hartwig JH, Falet H, Hoffmeister KM. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med 2015;21:47–54.
- Hsu AP, Davis J, Puck JM, Holland SM, Freeman AF. Autosomal dominant hyper IgE syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. 2010 Feb 23 [updated 2012 Jun 7].
- Mogensen TH. STAT3 and the Hyper-IgE syndrome: clinical presentation, genetic origin, pathogenesis, novel findings and remaining uncertainties. Jakstat 2013;2:e23435.
- Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007;448:1058–1062.
- Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 2007;357:1608–1619.
- Van De Veerdonk FL, Marijnissen RJ, Joosten LA, Kullberg BJ, Drenth JP, Netea MG, Van Der Meer JW. Milder clinical hyperimmunoglobulin E syndrome phenotype is associated with partial interleukin-17 deficiency. Clin Exp Immunol 2010;159:57–64.
- Zhang J, Wu J, Peng X, Song J, Wang J, Dong W. Associations between STAT3 rs744166 polymorphisms and susceptibility to ulcerative colitis and Crohn’s disease: a meta-analysis. PLoS One 2014;9:e109625.
- Ferguson LR, Han DY, Fraser AG, Huebner C, Lam WJ, Morgan AR, Duan H, Karunasinghe N. Genetic factors in chronic inflammation: single nucleotide polymorphisms in the STAT-JAK pathway, susceptibility to DNA damage and Crohn’s disease in a New Zealand population. Mutat Res 2010;690:108–115.
- Kotkowska A, Sewerynek E, Domanska D, Pastuszak-Lewandoska D, Brzezianska E. Single nucleotide polymorphisms in the STAT3 gene influence AITD susceptibility, thyroid autoantibody levels, and IL6 and IL17 secretion. Cell Mol Biol Lett 2015;20:88–101.
- Netea MG, Joosten LA, Li Y, Kumar V, Oosting M, Smeekens S, Jaeger M, Ter Horst R, Schirmer M, Vlamakis H, et al. Understanding human immune function using the resources from the human functional genomics project. Nat Med 2016;22:831–833.
- Ter Horst R, Jaeger M, Smeekens SP, Oosting M, Swertz MA, Li Y, Kumar V, Diavatopoulos DA, Jansen AF, Lemmers H, et al. Host and environmental factors influencing individual human cytokine responses. Cell 2016;167:1111-1124:e1113.
- Li Y, Oosting M, Deelen P, Ricano-Ponce I, Smeekens S, Jaeger M, Matzaraki V, Swertz MA, Xavier RJ, Franke L, et al. Inter-individual variability and genetic influences on cytokine responses to bacteria and fungi. Nat Med 2016;22:952–960.
- Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 2007;282:9358–9363.
- Shah TS, Liu JZ, Floyd JA, Morris JA, Wirth N, Barrett JC, Anderson CA. optiCall: a robust genotype-calling algorithm for rare, low-frequency and common variants. Bioinformatics 2012;28:1598–1603.
- Semple JW, Italiano JE Jr., Freedman J. Platelets and the immune continuum. Nat Rev Immunol 2011;11:264–274.
- Wu D, Xie J, Wang X, Zou B, Yu Y, Jing T, Zhang S, Zhang Q. Micro-concentration lipopolysaccharide as a novel stimulator of megakaryocytopoiesis that synergizes with IL-6 for platelet production. Sci Rep 2015;5:13748.
- Kaser A, Brandacher G, Steurer W, Kaser S, Offner FA, Zoller H, Theurl I, Widder W, Molnar C, Ludwiczek O, et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: role in inflammatory thrombocytosis. Blood 2001;98:2720–2725.
- Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, et al. Function of mitochondrial STAT3 in cellular respiration. Science 2009;323:793–797.
- Visavadiya NP, Keasey MP, Razskazovskiy V, Banerjee K, Jia C, Lovins C, Wright GL, Hagg T. Integrin-FAK signaling rapidly and potently promotes mitochondrial function through STAT3. Cell Commun Signal 2016;14:32.
- Woollard KJ, Suhartoyo A, Harris EE, Eisenhardt SU, Jackson SP, Peter K, Dart AM, Hickey MJ, Chin-Dusting JP. Pathophysiological levels of soluble P-selectin mediate adhesion of leukocytes to the endothelium through Mac-1 activation. Circ Res 2008;103:1128–1138.
- Chandesris MO, Azarine A, Ong KT, Taleb S, Boutouyrie P, Mousseaux E, Romain M, Bozec E, Laurent S, Boddaert N, et al. Frequent and widespread vascular abnormalities in human signal transducer and activator of transcription 3 deficiency. Circ Cardiovasc Genet 2012;5:25–34.
- Dutzmann J, Daniel JM, Bauersachs J, Hilfiker-Kleiner D, Sedding DG. Emerging translational approaches to target STAT3 signalling and its impact on vascular disease. Cardiovasc Res 2015;106:365–374.
- Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012;380:2095–2128.
- Wake MS, Watson CJ. STAT3 the oncogene - still eluding therapy? Febs J 2015;282:2600–2611.
- Huang W, Dong Z, Chen Y, Wang F, Wang CJ, Peng H, He Y, Hangoc G, Pollok K, Sandusky G, et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene 2016;35:783–792.
- Oh DY, Lee SH, Han SW, Kim MJ, Kim TM, Kim TY, Heo DS, Yuasa M, Yanagihara Y, Bang YJ. Phase I study of OPB-31121, an oral STAT3 inhibitor, in patients with advanced solid tumors. Cancer Res Treat 2015;47:607–615.
- Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, Harrison CN, Pane F, Zachee P, Mesa R, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med 2015;372:426–435.
- Goel D. Ticagrelor: the first approved reversible oral antiplatelet agent. Int J Appl Basic Med Res 2013;3:19–21.
- Marczewski MM, Postula M, Kosior D. Novel antiplatelet agents in the prevention of cardiovascular complications–focus on ticagrelor. Vasc Health Risk Manag 2010;6:419–429.
- Savi P, Pereillo JM, Uzabiaga MF, Combalbert J, Picard C, Maffrand JP, Pascal M, Herbert JM. Identification and biological activity of the active metabolite of clopidogrel. Thromb Haemost 2000;84:891–896.
- Abdulmalak C, Cottenet J, Beltramo G, Georges M, Camus P, Bonniaud P, Quantin C. Haemoptysis in adults: a 5-year study using the French nationwide hospital administrative database. Eur Respir J 2015;46:503–511.
- Lee BR, Yu JY, Ban HJ, Oh IJ, Kim KS, Kwon YS, Kim YI, Kim YC, Lim SC. Analysis of patients with hemoptysis in a tertiary referral hospital. Tuberc Respir Dis (Seoul) 2012;73:107–114.
- Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood 2003;101:2940–2954.
- Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, Miller JA, O’Connell AC, Puck JM. Hyper-IgE syndrome with recurrent infections–an autosomal dominant multisystem disorder. N Engl J Med 1999;340:692–702.