
Abstract
Thrombosis is a frequent issue in cancer patients. Tumor-induced platelet activation and coagulation does not only constitute a significant risk for thrombosis, but also contribute to tumor progression by promoting critical processes such as angiogenesis and metastasis. In addition to their role in hemostasis, platelets are increasingly recognized as regulators of inflammation. By modulating the immune system, platelets regulate several aspects of cancer-associated pathology. Platelets influence the inflammatory response in cancer by affecting the activation status of the endothelium and by recruiting leukocytes to primary and metastatic tumor sites, as well as to distant organs unaffected by tumor growth. Furthermore, platelets participate in the formation of neutrophil extracellular traps, which can promote metastasis, thrombosis, and contribute to organ failure. In this review, we discuss the role of platelets as coordinators of the immune system during malignant disease and the potential of targeting platelets to prevent cancer-associated pathology.
Platelets in cancer
Platelets, anucleated cells derived from megakaryocytes, are recognized for their essential role in hemostasis. When a wound is formed, platelets adhere to the vessel wall through specific cell surface–ligand interactions, which results in platelet activation, aggregation through fibrinogen bridging, and the formation of a primary hemostatic plug. Thrombin, generated through activation of the coagulation cascade, subsequently cleaves fibrinogen to form a stable fibrin clot that prevents bleeding and contributes to the healing processes. This function of platelets is critical to prevent us from bleeding to death upon injury. However, aberrant platelet activation is known to contribute to diseases such as thrombosis, and it is therefore important that the process of platelet activation is strictly regulated.
Tumors have been described as wounds that do not heal (Citation1). For example, tumors are characterized by constitutive angiogenesis and inflammation. Stroma production and infiltration of fibroblasts are other similarities between tumors and wounds.
Individuals with cancer have an increased risk for thrombotic events, and thrombosis is a major contributor to morbidity and mortality in cancer patients (Citation2). Several factors contribute to the pro-thrombotic state associated with malignant disease (Citation3). The leaky tumor vasculature enables fibrinogen leakage into the extravascular space, which in the presence of thrombin, expressed for instance by tumor cells, can be cleaved to form a fibrin clot. Moreover, clotting factor expression is often altered in the presence of a tumor. Expression of pro-coagulant factors such as tissue factor (TF), the inducer of the extrinsic coagulation pathway, and cancer procoagulant (CP), is frequently enhanced, and levels of anticoagulant factors such as tissue plasminogen activator are reduced. In addition to this, platelet activation and turnover is often increased in individuals with cancer. The tumor vessel leakiness contributes to platelet activation in the tumor due to exposure of subendothelial factors such as collagen and TF. Furthermore, aberrant expression of TF and CP by tumor cells and the tumor microenvironment contributes to activation of platelets (Citation2). High platelet counts are considered a poor prognostic factor in many cancer types, and a high platelet-lymphocyte ratio has been suggested to predict poor patient outcome (Citation4–Citation8).
Besides contributing to thrombosis, activated platelets can promote tumor progression. Upon activation, platelets release a plethora of factors that modulate the tumor microenvironment. Pro-angiogenic growth factors such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) are released from alpha-granules of activated platelets and stimulate tumor angiogenesis. Release of pro-inflammatory cytokines drives recruitment of leukocytes, which contribute to remodeling of the extracellular matrix and the angiogenic process. Platelets are also recognized as promoters of metastasis via several distinct mechanisms. Platelets can enhance epithelial to mesenchymal transition and tumor cell survival in the circulation, extravasation, and colonization at the secondary site (Citation9,Citation10). The importance of platelet support to enable establishment of metastases in secondary organs is illustrated by numerous studies in mice, revealing that inhibition of platelet activation efficiently suppresses metastasis (Citation9,Citation11–Citation13).
While the role of platelets in hemostasis and thrombosis has been known for long, platelets have more recently emerged as critical regulators of inflammation. This review will focus on the role of platelets as inflammatory regulators in cancer and their impact on tumor progression and cancer-associated systemic disease ().
Figure 1. The pro-inflammatory roles of platelets in cancer. Platelets contribute to cancer-associated inflammation in several ways. Enhanced platelet activation in the presence of a tumor elevates the risk for thrombosis (left panel). Intravascular thrombi may cause vessel occlusion, impair vascular perfusion, and cause endothelial activation with upregulation of adhesion molecules such as E-selectin, P-selectin, ICAM, and VCAM. These receptors mediate leukocyte extravasation into the tissue but can also directly or indirectly promote extravasation of cancer cells. Activated platelets degranulate and secrete pro-inflammatory factors, such as CXCL5 and CXCL7, contributing to leukocyte recruitment (middle panel). Platelets can promote formation of neutrophil extracellular traps (NETs) and associated immunothrombosis via release of platelet granule cytokines, as well as direct contact with neutrophils via cell surface receptors (right panel). NETs contribute to systemic effects of cancer by promoting metastasis, thrombosis, and organ failure.
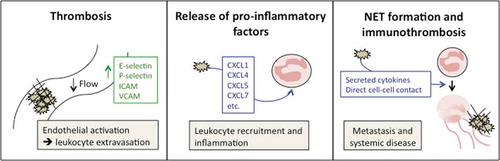
Thrombosis contributes to vascular inflammation
A link between cancer and thrombosis was recognized already in the mid-nineteenth century, when Armand Trosseau discovered that thrombotic events were frequently caused by presence of occult tumors (Citation14). It is nowadays well known that cancer patients have an elevated risk for thrombosis and that this significantly contributes to cancer-associated death (Citation15).
Thrombi formation is one potential cause of inflammation in distant organs of cancer patients. A thrombus in the microvasculature will cause vessel occlusion and prevent normal blood flow. Reduced vascular perfusion will be sensed by the vessel wall and result in activation of the endothelium—a process that promotes inflammation. Upon endothelial activation, adhesion molecules responsible for capture and extravasation of leukocytes will be expressed. In a stepwise process, these endothelial receptors will allow leukocytes, such as neutrophils, to adhere to the endothelium, migrate through the vessel wall, and enter the tissue (Citation16). The first response takes place only within minutes after endothelial activation is induced, when preformed P-selectin stored in Weibel Palade-bodies is released from endothelial cells and displayed on the endothelial surface (Citation17). Leukocytes bind transiently and weak to selectins on the endothelium via P-selectin glycoprotein ligand-1 (PSGL-1) on their surface. In parallel, de novo formation of both P- and E-selectin is induced, together with integrin receptors such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). These CAMs are responsible for firm adhesion of the leukocytes, by interacting with integrins such as lymphocyte function associated antigen-1 (LFA-1) and macrophage-1 antigen (Mac-1). The integrin-mediated interactions will also induce signaling events in the endothelial cells leading to loosening of the endothelial junctions, facilitating transendothelial migration, and transcription of additional pro-inflammatory factors. The infiltrating immune cells will produce and secrete pro-inflammatory factors that will recruit more leukocytes to the site and hence exacerbate the inflammation. However, it may be worth to notice that platelets themselves actively participate in capturing of leukocytes. Activated platelets do also express receptors on their surface that allows direct interactions with immune cells, such as P-selectin and CD40L (Citation18,Citation19). A study by Sreeramkumar et al. exemplifies the significance of platelet P-selectin and leukocyte PSGL-1 in inflammation (Citation20). It was shown that neutrophils that interact with activated endothelium polarize and protrude the trailing end of the cell body, the uropod, into the vessel lumen. The uropod contains PSGL-1 clusters that can “catch” activated platelets in the circulation expressing P-selectin. When this interaction takes place, the neutrophil is able to organize additional receptors needed for transmigration of the endothelium and initiate inflammation (Citation20).
Several studies also indicate that endothelial activation promotes metastasis (Citation21). Recruitment and transmigration of leukocytes into the tissue is likely an important factor for this effect. Formation of pre-metastatic niches have been described, where VEGFR1+ leukocytes prepare the “soil” for incoming metastatic tumor cells (Citation22). Hypothetically, endothelial activation in distant organs due to thrombosis could directly promote extravasation of tumor cells. It has been described that selectins expressed by activated endothelial cells can be used as a transmigration route also by extravasating tumor cells (Citation23–Citation25). In this way, vascular inflammation induced by thrombosis could stimulate tumor cell entry into tissues of distant organs.
Secretion of pro-inflammatory factors from platelets contribute to tumor progression and inflammation
Leukocytes can make up a significant amount of the total cell count in the primary tumor. Macrophages, for example, are highly abundant in the tumor microenvironment and affect various processes during malignant progression from tumor initiation to metastasis (Citation26). Neutrophils are often less numerous but are nonetheless critical modulators of tumor development and progression (Citation27). For example, neutrophils can mediate the initial angiogenic switch in a mouse model of pancreatic neuroendocrine tumorigenesis, an effect attributed to secretion of the matrix metalloproteinase 9 (Citation28).
Platelets contain several pro-inflammatory factors that are released upon platelet activation, such as CXCL1, CXCL4, CXCL5, CXCL7, CXCL12 (SDF-1), and interleukin (IL)-8 (Citation29). These factors are potent recruiters and activators of leukocytes like neutrophils and monocytes. However, these molecules are not only produced by platelets and pro-inflammatory factors produced by various cell types can contribute to leukocyte recruitment. Besides these preformed factors that are immediately released upon platelet degranulation, platelet activation results in synthesis of IL-1β from a pre-mRNA stored in the platelet (Citation30). IL-1β upregulates expression of endothelial leukocyte adhesion molecules and induces TF expression on the endothelial cell surface, thereby contributing to platelet activation and inflammation (Citation31). Furthermore, IL-1β promotes further platelet activation in an autocrine manner by signaling via the IL-1R on platelets (Citation32). Moreover, platelets constitute a major source of transforming growth factor (TGF)-β in the circulation (Citation33). Enhanced platelet activation results in increased release of TGF-β, which can promote tumorigenesis in different ways. TGF-β is a potent immunosuppressive cytokine and elevated expression in tumors promotes escape from recognition and killing of tumor cells by the immune system (Citation34).
How leukocytes impact tumor development and progression is a complex issue, which has led to attempts to divide both macrophages and neutrophils into subgroups with different properties and effects on tumor progression. For macrophages, a division into M1- and M2-macrophages has been established, with M1-macrophages having a tumor-suppressive phenotype, while M2-macrophages, on the other hand, are tumor promoting (Citation35). Such a categorization is, however, now regarded as too simplistic and a spectrum of macrophage phenotypes is probably more likely than a strict polarization into anti- or pro-tumorigenic. A similar attempt to divide neutrophils into N1- and N2-neutrophils has been made, but also here the situation is likely to be more complex. Undoubtedly, leukocytes in the tumor microenvironment are more plastic than previously assumed. One factor implicated in pushing the neutrophils toward a pro-tumorigenic phenotype is TGF-β (Citation36). Platelets are a rich source of TGF-β (Citation37), and platelets activated by the tumor will contribute to the TGF-β levels in the microenvironment.
Leukocytes are not only involved in modulation of the tumor microenvironment but also contribute to metastasis. Numerous studies reveal that both neutrophils and macrophages support survival and growth of metastatic tumor cells, for example, by promoting angiogenesis. The concept of pre-metastatic niches was presented about a decade ago, suggesting that primary tumors prepare the environment in distant organs to support growth and survival of incoming metastatic tumor cells (Citation38). The niche formation is orchestrated to a large extent by leukocytes, which are recruited to the distant sites by soluble pro-inflammatory factors produced by the primary tumor. Recent data now suggest that platelets may have a crucial role for leukocyte recruitment to pre-metastatic niches. A study published a few years ago revealed that interaction between platelets and tumor cells in the vasculature was required for recruitment of neutrophils to the metastatic site (Citation39). The neutrophils, essential for metastasis to form, were recruited to the site by CXCL5 and CXCL7, secreted by platelets upon contact with tumor cells. Platelet depletion completely suppressed neutrophil recruitment to the metastatic site, which strongly indicates that platelets have a critical role as neutrophil attractants. Platelets are also a rich source of CXCL12 that function as a chemotactic factor to recruit CXCR4-positive cells such as macrophages to the metastatic site (Citation40).
Platelets can induce production of pro-inflammatory factors from other cells in the tumor microenvironment and in this way indirectly promote recruitment of leukocytes to primary and metastatic tumors. For example, platelets have been shown to induce CCL2 expression in tumor cells, and CCL2 can recruit monocytes that promote metastatic seeding at distant sites (Citation10,Citation41–Citation43). Similarly, platelets, together with leukocytes and tumor cells, induced CCL5 secretion from endothelial cells in an experimental model of colorectal cancer, which enhanced metastatic seeding due to recruitment of monocytes (Citation44).
We are not aware of studies directly connecting thrombocytopenia or thrombocytosis to modulation of inflammation in cancer patients. There are however other inflammatory diseases such as atherosclerosis, where a connection to platelet chemokines has been demonstrated (Citation45).
Tumor-induced NET formation: a potential role for platelets?
Next to their role as leukocyte recruiters, platelets have more recently been implicated in the formation of neutrophil extracellular traps (NETs). Formation of NETs was first described in 2004 as an innate immune defense mechanism used to combat severe bacterial infections (Citation46). During NET formation, activated neutrophils release their chromatin and granular content, resulting in the formation of web-like structures made up of extracellular DNA strands with associated proteins such as histones and neutrophil proteases. This web can physically trap pathogens, while the local high concentration of antimicrobial agents in the NETs enables efficient killing of the bacteria. The initial finding of bacterial-induced NET formation has been followed by numerous studies, describing a role for NETs also in sterile inflammation, for example in cancer (Citation47,Citation48). NETs were first detected in mice with cancer (Citation49), but were soon thereafter found in biopsies from human tumors (Citation50).
While formation of NETs may be beneficial to fight an infection, this process is also a threat to the individual since it is associated with vascular damage (Citation51–Citation53). Moreover, NETs have a pro-thrombotic effect since extracellular DNA activates platelets (Citation54). NETs also promote binding and activation of factor XII, inducer of the intrinsic coagulation pathway (Citation55). In this way, netting neutrophils promote thrombotic disease, a phenomenon sometimes referred to as immunothrombosis. Despite the apparent disadvantages of NET formation for the host, formation of NETs may be a last resort to protect the individual from life-threatening sepsis during a severe infection. This fine edge between benefit and risk when it comes to NET formation indicates that a strict regulation is needed to ensure that NETs are not formed if not necessary. During infection, platelets have been identified as such regulators by sensitizing the level of bacteria-derived lipopolysaccharide (LPS). Platelets bind LPS via Toll-like receptor 4 on their surface and induce NET formation in neutrophils only when the LPS concentration has reached a critical level (Citation56). Furthermore, interaction between P-selectin, soluble or on the platelet surface, and PSGL-1 on neutrophils, was shown to promote NET generation in murine neutrophils (Citation57). Several other reports further support a critical role of platelets as regulators of NET formation in different conditions (Citation58).
So why are NETs formed in cancer patients and what are the consequences? Two tumor-derived factors that have been identified as inducers of NETs are granulocyte colony-stimulating factor (G-CSF) and IL-8 (Citation49,Citation59,Citation60). If platelets play a role also in tumor-induced NET formation is currently not known, but not unlikely considering the elevated platelet activation in individuals with cancer. It was recently suggested that platelets prime tumor cells to promote NET formation (Citation33). The authors suggest that this effect is due to platelets acting as carriers of tumor-derived exosomes, which in turn promote NET generation. A role for tumor-derived exosomes in the formation of NETs is also supported by another recent study, but platelets were not considered as carriers of the exosomes in this report (Citation61).
NETs have been implicated in tumor progression and metastasis (Citation62–Citation64), but also in systemic effects associated with cancer. These systemic effects are often connected to the pro-thrombotic role of NETs. NETs have been shown to promote thrombosis both in tumor-bearing mice and in cancer patients (Citation49,Citation61,Citation65). Moreover, NETs in association with platelets contribute to kidney injury in mice with cancer (Citation60,Citation66). This effect was associated with vessel occlusion and could be suppressed by removal of NETs by treatment with DNase I or by prevention of de novo NET formation using an inhibitor of the enzyme peptidylarginine deiminase 4 (PAD4).
While accumulating data support that platelets induce NET formation during infection, more solid support is needed to clarify which importance platelets have in tumor-associated NET formation.
Activated platelets as therapeutic targets to treat cancer
The role of platelets in regulation of cancer-associated inflammation and thrombosis suggests that tumor-induced platelet activation is a potential therapeutic target. However, such approaches would have to be carefully designed to avoid risk for bleedings, considering the importance of platelets as regulators of hemostasis. Low-molecular-weight heparin is used in the clinic to treat cancer-associated thrombosis (Citation67). A direct anticancer function of heparin has been suggested, but the relevance of such effects has been debated (Citation68,Citation69). Several studies have recently reported an anticancer effect of aspirin (Citation70–Citation72). Aspirin has been in clinical use for more than a century to treat patients with coagulopathies such as acute coronary syndrome. Acetylsalicylic acid, the functional substance of aspirin, inhibits COX-enzymes involved in generation of thromboxane A2, a potent inducer of platelet activation and aggregation. A protective effect of aspirin in the preventive setting, especially for colorectal cancer, was discussed already in the late twentieth century (Citation73). In 2016, a study including more than 130 000 persons and with a follow-up time of more than 30 years revealed that regular use of aspirin was associated with significantly decreased risk for developing cancer, especially tumors of the gastrointestinal tract (Citation74). The antitumor effect of aspirin seems to be achieved with doses of aspirin around 75 mg/day, with no further therapeutic benefit if the dose is increased. This indicates that regular use of aspirin at low doses, associated with limited risks for unwanted side effects such as bleeding, could be a strategy to prevent cancer. A number of clinical trials are currently ongoing to evaluate the therapeutic effect of aspirin treatment in cancer patients (Citation75). Whether the effects of aspirin in cancer patients are platelet-dependent is however still not clear and warrants further investigation. Another potential strategy to suppress platelet activation is to target ADP-mediated platelet activation with inhibitors of the P2Y12 receptor, such as clopidogrel. Several studies indicate that such therapeutic approaches, alone or in combination with aspirin, could be effective to treat cancer (Citation76).
To conclude, it is clear that inflammatory factors impact on tumor development and progression. Platelets have emerged as potent influencers of cancer-associated inflammation as they are involved in endothelial activation, recruitment of leukocytes, and formation of NETs. To limit platelet activation in cancer patients is therefore a relevant aspect to take into account when designing new therapeutic approaches to treat cancer.
Declaration of interest
The authors report no conflicts of interest.
References
- Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–1659. doi:10.1056/NEJM198612253152606.
- Ruf W. Tissue factor and cancer. Thromb Res. 2012;130(Suppl 1):SS84–7. doi:10.1016/j.thromres.2012.08.285.
- Lip GY, Chin BS, Blann AD. Cancer and the prothrombotic state. Lancet Oncol. 2002;3(1):27–34. doi:10.1016/S1470-2045(01)00619-2.
- Chen N, et al. Increased platelet-lymphocyte ratio closely relates to inferior clinical features and worse long-term survival in both resected and metastatic colorectal cancer: an updated systematic review and meta-analysis of 24 studies. Oncotarget. 2017;8(19):32356–32369.
- Gu X, et al. Clinicopathological and prognostic significance of platelet to lymphocyte ratio in patients with gastric cancer. Oncotarget. 2016;7(31):49878–49887. doi:10.18632/oncotarget.10490.
- Wang J, et al. Prognostic role of pretreatment platelet to lymphocyte ratio in urologic cancer. Oncotarget. 2017;8(41):70874–70882.
- You J, et al. Preoperative platelet to lymphocyte ratio is a valuable prognostic biomarker in patients with colorectal cancer. Oncotarget. 2016;7(18):25516–25527. doi:10.18632/oncotarget.8334.
- Zhang M, et al. High platelet-to-lymphocyte ratio predicts poor prognosis and clinicopathological characteristics in patients with breast cancer: A meta-analysis. Biomed Res Int. 2017;2017:9503025. doi:10.1155/2017/9503025.
- Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11(2):123–134. doi:10.1038/nrc3004.
- Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20(5):576–590. doi:10.1016/j.ccr.2011.09.009.
- Gasic GJ, Gasic TB, Stewart CC. Antimetastatic effects associated with platelet reduction. Proc Natl Acad Sci. 1968;61(1):46–52. doi:10.1073/pnas.61.1.46.
- Nieswandt B, et al. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999;59(6):1295–1300.
- Palumbo JS, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. 2005;105(1):178–185. doi:10.1182/blood-2004-06-2272.
- Trousseau A. Plegmasia alba dolens. Lectures on clinical medicine, delivered at the Hotel-Dieu, Paris. 1865;5:281–332.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood. 2007;110(6):1723–1729. doi:10.1182/blood-2006-10-053736.
- Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. 2014;41(5):694–707. doi:10.1016/j.immuni.2014.10.008.
- Bevilacqua MP, Nelson RM. Selectins. J Clin Invest. 1993;91(2):379–387. doi:10.1172/JCI116210.
- Lievens D, et al. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood. 2010;116(20):4317–4327. doi:10.1182/blood-2010-01-261206.
- Thomas MR, Storey RF. The role of platelets in inflammation. Thromb Haemost. 2015;114(3):449–458. doi:10.1160/TH14-12-1067.
- Sreeramkumar V, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. 2014;346(6214):1234–1238. doi:10.1126/science.1256478.
- Matsuo Y, et al. Involvement of p38alpha mitogen-activated protein kinase in lung metastasis of tumor cells. J Biol Chem. 2006;281(48):36767–36775. doi:10.1074/jbc.M604371200.
- Peinado H, et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat Rev Cancer. 2017;17(5):302–317. doi:10.1038/nrc.2017.6.
- Aigner S, et al. CD24, a mucin-type glycoprotein, is a ligand for P-selectin on human tumor cells. Blood. 1997;89(9):3385–3395.
- Brodt P, et al. Liver endothelial E-selectin mediates carcinoma cell adhesion and promotes liver metastasis. Int J Cancer. 1997;71(4):612–619. doi:10.1002/(SICI)1097-0215(19970516)71:4<612::AID-IJC17>3.0.CO;2-D.
- Iwai K, et al. Importance of E-selectin (ELAM-1) and sialyl Lewis(a) in the adhesion of pancreatic carcinoma cells to activated endothelium. Int J Cancer. 1993;54(6):972–977. doi:10.1002/ijc.2910540618.
- Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. doi:10.1016/j.immuni.2014.06.010.
- Coffelt SB, Wellenstein MD, De Visser KE. Neutrophils in cancer: nNeutral no more. Nat Rev Cancer. 2016;16(7):431–446. doi:10.1038/nrc.2016.52.
- Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci. 2006;103(33):12493–12498. doi:10.1073/pnas.0601807103.
- Gleissner CA, Von Hundelshausen P, Ley K. Platelet chemokines in vascular disease. Arterioscler Thromb Vasc Biol. 2008;28(11):1920–1927. doi:10.1161/ATVBAHA.108.169417.
- Lindemann S, et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. 2001;154(3):485–490. doi:10.1083/jcb.200105058.
- Bevilacqua MP, et al. Interleukin-1 activation of vascular endothelium. Effects on procoagulant activity and leukocyte adhesion. Am J Pathol. 1985;121(3):394–403.
- Brown GT, et al. Lipopolysaccharide stimulates platelets through an IL-1beta autocrine loop. J Immunol. 2013;191(10):5196–5203. doi:10.4049/jimmunol.1300354.
- Abdol Razak N, Elaskalani O, Metharom P. Pancreatic cancer-induced neutrophil extracellular traps: a potential contributor to cancer-associated thrombosis. Int J Mol Sci. 2017;18(3). doi:10.3390/ijms18030487.
- Neuzillet C, et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther. 2015;147:22–31. doi:10.1016/j.pharmthera.2014.11.001.
- Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. 2014;5:514. doi:10.3389/fimmu.2014.00514.
- Fridlender ZG, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3):183–194. doi:10.1016/j.ccr.2009.06.017.
- Assoian RK, et al. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem. 1983;258(11):7155–7160.
- Kaplan RN, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–827. doi:10.1038/nature04186.
- Labelle M, Begum S, Hynes RO. Platelets guide the formation of early metastatic niches. Proc Natl Acad Sci. 2014;111(30):E3053–61. doi:10.1073/pnas.1411082111.
- Sun X, et al. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010;29(4):709–722. doi:10.1007/s10555-010-9256-x.
- Palframan RT, et al. Inflammatory chemokine transport and presentation in HEV: A remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med. 2001;194(9):1361–1373. doi:10.1084/jem.194.9.1361.
- Qian BZ, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. doi:10.1038/nature10138.
- Wolf MJ, et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell. 2012;22(1):91–105. doi:10.1016/j.ccr.2012.05.023.
- Laubli H, Spanaus KS, Borsig L. Selectin-mediated activation of endothelial cells induces expression of CCL5 and promotes metastasis through recruitment of monocytes. Blood. 2009;114(20):4583–4591. doi:10.1182/blood-2008-10-186585.
- Blanchet X, et al. Inflammatory role and prognostic value of platelet chemokines in acute coronary syndrome. Thromb Haemost. 2014;112(6):1277–1287. doi:10.1160/TH14-02-0139.
- Brinkmann V, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi:10.1126/science.1092385.
- Cedervall J, Zhang Y, Olsson AK. Tumor-induced NETosis as a risk factor for metastasis and organ failure. Cancer Res. 2016;76(15):4311–4315. doi:10.1158/0008-5472.CAN-15-3051.
- Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23(3):279–287. doi:10.1038/nm.4294.
- Demers M, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci. 2012;109(32):13076–13081. doi:10.1073/pnas.1200419109.
- Berger-Achituv S, et al. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol. 2013;4:48. doi:10.3389/fimmu.2013.00048.
- Gupta AK, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010;584(14):3193–3197. doi:10.1016/j.febslet.2010.06.006.
- Saffarzadeh M, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012;7(2):e32366. doi:10.1371/journal.pone.0032366.
- Villanueva E, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187(1):538–552. doi:10.4049/jimmunol.1100450.
- Fuchs TA, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci. 2010;107(36):15880–15885. doi:10.1073/pnas.1005743107.
- von Bruhl ML, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819–835. doi:10.1084/jem.20112322.
- Clark SR, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–469. doi:10.1038/nm1565.
- Etulain J, et al. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015;126(2):242–246. doi:10.1182/blood-2015-01-624023.
- Carestia A, Kaufman T, Schattner M. Platelets: new bricks in the building of neutrophil extracellular traps. Front Immunol. 2016;7:271. doi:10.3389/fimmu.2016.00271.
- Alfaro C, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res. 2016;22(15):3924–3936. doi:10.1158/1078-0432.CCR-15-2463.
- Cedervall J, et al. Neutrophil extracellular traps accumulate in peripheral blood vessels and compromise organ function in tumor-bearing animals. Cancer Res. 2015;75(13):2653–2662. doi:10.1158/0008-5472.CAN-14-3299.
- Leal AC, et al. Tumor-derived exosomes induce the formation of neutrophil extracellular traps: implications for the establishment of cancer-associated thrombosis. Sci Rep. 2017;7(1):6438. doi:10.1038/s41598-017-06893-7.
- Cools-Lartigue J, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013;123:3446–3458. doi:10.1172/JCI67484.
- Park J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8(361):361ra138. doi:10.1126/scitranslmed.aag1711.
- Tohme S, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016;76(6):1367–1380. doi:10.1158/0008-5472.CAN-15-1591.
- Thalin C, et al. NETosis promotes cancer-associated arterial microthrombosis presenting as ischemic stroke with troponin elevation. Thromb Res. 2016;139:56–64. doi:10.1016/j.thromres.2016.01.009.
- Cedervall J, et al. Pharmacological targeting of peptidylarginine deiminase 4 prevents cancer-associated kidney injury in mice. Oncoimmunology. 2017;6(8):e1320009. doi:10.1080/2162402X.2017.1320009.
- Elalamy I, et al. Long-term treatment of cancer-associated thrombosis: The choice of the optimal anticoagulant. J Thromb Haemost. 2017;15(5):848–857. doi:10.1111/jth.13659.
- Barni S, et al. The effect of low-molecular-weight heparin in cancer patients: The mirror image of survival? Blood. 2014;124(1):155–156. doi:10.1182/blood-2014-03-561761.
- Zhang N, et al. Low molecular weight heparin and cancer survival: clinical trials and experimental mechanisms. J Cancer Res Clin Oncol. 2016;142(8):1807–1816. doi:10.1007/s00432-016-2131-6.
- Gresele P, et al. Platelet-targeted pharmacologic treatments as anti-cancer therapy. Cancer Metastasis Rev. 2017;36:331–355. doi:10.1007/s10555-017-9679-8.
- Rothwell PM, et al. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011;377(9759):31–41. doi:10.1016/S0140-6736(10)62110-1.
- Rothwell PM, et al. Effect of daily aspirin on risk of cancer metastasis: Astudy of incident cancers during randomised controlled trials. Lancet. 2012;379(9826):1591–1601. doi:10.1016/S0140-6736(12)60209-8.
- Marnett LJ. Aspirin and the potential role of prostaglandins in colon cancer. Cancer Res. 1992;52(20):5575–5589.
- Cao Y, et al. Population-wide impact of long-term use of aspirin and the risk for cancer. JAMA Oncol. 2016;2(6):762–769. doi:10.1001/jamaoncol.2015.6396.
- Santilli F, Boccatonda A, Davi G. Aspirin, platelets, and cancer: The point of view of the internist. Eur J Intern Med. 2016;34:11–20. doi:10.1016/j.ejim.2016.06.004.
- Elaskalani O, et al. Targeting platelets for the treatment of cancer. Cancers (Basel). 2017;9(7). doi:10.3390/cancers9070094.