
Abstract
Introduction
Platelets are critical for hemostasis, and a low platelet count predicts mortality in trauma. The role of platelet dysfunction in severe traumatic hemorrhage and coagulopathy needs to be further defined. The aim of this study was to evaluate the platelet function in a new model of experimental traumatic hemorrhage.
Material and methods
New Zealand white rabbits (n = 10) were subjected to tracheostomy and trauma laparotomy, and then bilateral femur fractures with 40% hemorrhage of their estimated blood volume. Arterial blood gases, standard coagulation tests, mean platelet volume, platelet aggregation using impedance aggregometry with agonist collagen, arachidonic acid (ASPI), and adenosine diphosphate (ADP), rotational thromboelastometry, and fibrinogen binding of platelets were analyzed using flow cytometry.
Results
After traumatic hemorrhage, there was a significant physiological response with a rise in lactate (P < .001) and a decrease in base excess (P < .001) and temperature (P < .001). Platelet count decreased from a mean of 244x109/L to 94 x109/L (P = .004) and the mean platelet volume increased from 5.1fL to 6.1fL (P = .002). Impedance aggregometry with the agonist collagen, ASPI, and ADP was all significantly decreased after hemorrhage (P = .007). However, there was an increased fibrinogen binding of ADP-activated platelets after traumatic hemorrhage analyzed by flow cytometry (P < .05).
Conclusions
This traumatic hemorrhage model presents two parallel pathophysiological responses of platelets; platelet consumption as evidenced by a significant decrease in platelet count and aggregation, and platelet hyperreactivity as shown by a higher mean platelet volume and enhanced platelet fibrinogen binding. Further studies are needed to characterize these different aspects of platelet function in severe traumatic hemorrhage.
Introduction
Major trauma is the leading cause of death worldwide in the young population. Hemorrhage is the most common preventable cause of death after trauma and accounts for up to 30%-40% of trauma-related deaths [Citation1–Citation3]. Massive hemorrhage in combination with tissue injury in trauma leads to a coagulopathic state, so-called trauma-induced coagulopathy (TIC), which is present in 25%-37% of the patients [Citation4]. Patients hospitalized with TIC have a significantly higher mortality risk than those without coagulation anomalies[Citation5]. Several factors are affected in TIC; different degrees of dysfibrinogenemia, hyperfibrinolysis, endothelial dysfunction, and impaired platelet function have all been described [Citation6]. Platelets play critical roles in hemostasis, and a low platelet count predicts mortality in trauma [Citation7]. Platelet dysfunction has been described as an early component in TIC, and previous studies have shown impaired platelet function both dependent or independent of platelet counts [Citation6,Citation8–Citation14].
Animal models of trauma hemorrhage and coagulopathy are important to further improve our knowledge in these conditions [Citation15]. The clinical relevance of some of these models has been discussed. The experimental rabbit model has previously been proven to be useful to simulate trauma hemorrhage [Citation16]. The pathophysiology of platelet dysfunction in severe traumatic hemorrhage and coagulopathy needs to be further studied. The purpose of this study was to evaluate platelet function in an experimental traumatic hemorrhage model.
Material and Methods
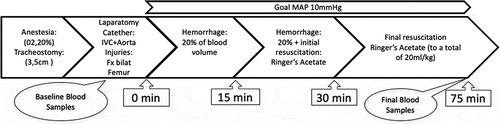
Ten New Zealand White Rabbits (3100–3800 g) were used in an experimental traumatic model with bilateral femur fractures, laparotomy and a class IV hemorrhage shock (>40% hemorrhage of blood volume) (). The study was approved (N67/15) by the Ethics Committee for Experiments in Animals, Karolinska Institutet, Stockholm, Sweden.
Figure 1. Timeline of the experimental traumatic hemorrhage model. Minute 0 simulates time of injury; the initial 15-min period simulates the time after trauma, waiting for transport, the second 15-min period simulates prehospital resuscitation during transportation, and the last 45 min simulates the initial treatment at the hospital with resuscitation.
Anesthesia
For premedication, medetomidine (0.2 mg/kg) was administered subcutaneously. Total intravenous anesthesia (TIVA) initially using sufentanil (6.9 mg/kg per hour) (Sufenta, Jansen-Cilag, Sweden), and midazolam (1.35 mg/kg per hour) (Actavis AB, Sweden) during laparotomy and femur fractures. Before initiation of hemorrhage and resuscitation, the TIVA was changed to intravenous ketamine infusion for the remainder of the experiment. No neuromuscular blocking agents were used. The animals were tracheotomized with a 3.5 mm cuffed endotracheal tube (Kimberly-Clark®) and started on mechanical ventilation with 25–30% oxygen in air using pressure-controlled mechanical ventilation, 8–15 breaths per minute with 4 cm H2O post end expiratory pressure (PEEP) and peak pressure of 20 cm H2O (Servo 900 C®, Siemens-Elema, Sweden).
Vascular Instrumentation
An arterial line was placed in the right ear artery for initial blood pressure monitoring and blood sampling. Percutaneous venous catheters were placed in both ears for the administration of drugs. After laparotomy, a 5 Fr introducer was placed, using a micropuncture technique, in the inferior vena cava (IVC) with the catheter tip in the right atrium of the heart for blood drainage. The abdominal aorta was cannulated in a similar fashion with an 8Fr Fem-Flex II arterial cannula (Edwards Lifesciences™). The catheter was used for blood sampling and continuous monitoring of the blood pressure.
Monitoring
The animals were monitored continuously with an electrocardiogram (ECG), mean arterial pressure (MAP) in the abdominal aorta, esophageal temperature, pulse oximetry on the tongue, and end-tidal CO2.
Traumatic Hemorrhage Phase
The rabbits initially sustained midline laparotomy and bilateral femur fractures. The femur fractures were standardized by stabilizing femur with two poly grip pliers right above the patella and 5-cm proximal of the patella. The fracture was then manually created between the pliers by outward rotation. A dislocation of the bone was confirmed in the closed compartment. Introducers (four French) were positioned in the IVC and the abdominal aorta. Ice packs were placed in the abdomen to rapidly cool down the animal and cause hypothermia aiming at 32°C. Forty percent of the rabbit's estimated blood volume (70 ml/kg) was drawn with syringes from the aorta to generate the equivalent of a class IV hemorrhagic shock. This was done by drawing blood from the aorta during the first 15 min, 20% of the blood volume, without resuscitation to simulate the immediate situation after trauma. The blood was manually withdrawn with a 20-ml syringe at a continuous rate. During the next 15 min, another 20% of the blood volume was drawn with a simultaneous infusion of Ringer's solution in the IVC, this phase was to simulate prehospital transportation and initial resuscitation. Thereafter, animals were kept in hemorrhagic shock for another 45 min. During the whole shock phase, animals were kept on a MAP-target of 10 mm Hg (pressure–targeted controlled hemorrhagic shock) and a target temperature of less than 32°C. Ringer´s solution was infused, corresponding to 40 ml/kg, to keep the MAP at 10 mm Hg. The total hemorrhagic shock time was 75 min ().
Blood Chemistry and Coagulation Status
Laboratory parameters of arterial blood (pH, pCO2, pO2, base excess (BE), hemoglobin (Hb), hematocrit (Hct), potassium (K+), sodium (Na+), calcium (Ca [Citation2]+), chloride (Cl−), glucose, and lactate) were monitored at baseline (0 min) and after 75 min of resuscitation or immediately if asystole. Blood samples were collected from the aortic catheter, at baseline and 75 min after trauma, using a vacutainer containing 1/10 volume of 3.8% sodium citrate. Fibrinogen concentration and prothrombin complex (PT)/international normalized ratio (INR) were also monitored both at baseline and 75 min after trauma.
Platelet Count and Mean Platelet Volume Measurement
Whole blood was 1:10 diluted with PBS. Platelet count and mean platelet volume (MPV) were measured by the blood cell analyzer ABX Micros 60 (ABX Diagnostics, Montpellier, France).
Rotational Thromboelastometry
ROTEM® is a viscoelastic method using whole blood collected in tubes containing 0.129-mol L-1 sodium citrate and analyzed as recommended by the manufacturer’s instructions ROTEM® Delta 3000 (GmbH, Munich, Germany). Samples were collected at baseline and 75 min after trauma. Analyses were performed within 30 min at a temperature adjusted to the actual rabbit body temperature. In ROTEM® the coagulation is displayed as a curve using the EXTEM reagent to illustrate the extrinsic pathway, the INTEM reagent for the intrinsic pathway and FIBTEM to show the fibrinogen function. The variables analyzed were EXTEM and INTEM clotting Time (CT) that indicates the time until a blood clot is initiated, and EXTEM and FIBTEM Maximum Clot Firmness (MCF) which is a value of the strength of the clot, with and without the platelet contribution. ROTEM® platelet is a module added to the device testing platelet aggregation by a change in impedance, similar to Multiplate. ROTEM® platelet was used to measure platelet aggregation in whole blood, platelet agonist adenosine diphosphate (ADP), the results were reported as the area under the curve (AUC) in Ohms*minutes.
Impedance Aggregometry
Impedance aggregometry (Multiplate®; Roche Diagnostics International Ltd, Switzerland) was used to test platelet function in whole blood by detecting a change in impedance as an effect of platelet aggregation on electrodes. Platelet aggregation was measured in whole blood at baseline and 75 min after trauma. Samples were prepared according to the manufacturer's recommendation. Platelet agonists were adenosine diphosphate (ADP), arachidonic acid (ASPI) and collagen (COL). Platelet responses to agonists were reported as the area under the aggregation curve (AUC) in units (u).
Flow Cytometry
Flow cytometry analysis of platelets was performed as previously described in the literature [Citation17,Citation18]. Briefly, 5 µl whole blood was added to 45 µl Tyrode’s-HEPES buffer containing appropriately diluted fluorescein isothiocyanate (FITC)–conjugated chicken anti-fibrinogen polyclonal antibody (product #2871; Diapensia HB, Linköping, Sweden) [Citation19] and in the presence of ADP (0, 0.3, 1, 10 μM, final concentrations). The samples were incubated for 20 min at room temperature, and mildly fixed and diluted with 1500 µl 0.5% formaldehyde saline. Flow cytometric analyses of rabbit platelet activation were carried out by monitoring platelet fibrinogen binding with a Beckman-Coulter FC500 flow cytometer (Beckman–Coulter Corp., Hialeah, FL, USA). Rabbit platelets were gated according to their characteristic light scattering signals. The latter is similar to human platelets that can be confirmed by platelet-specific marker CD41 (>98% positive; phycoerythrin/PE-conjugated CD41 monoclonal antibody clone HIP8; BD Biosciences, San Diego, CA, USA). Platelet fibrinogen binding was reported as the percentage of positive cells in the total platelet population.
Statistical Analysis
Data were presented as mean±SEMs if normally distributed; median and interquartile range (IQR) if non-normally distributed. Comparisons of platelet parameters before and after trauma were performed with Student paired t test using Microsoft Excel ® (Microsoft Corporation; Redmond, WA, USA) and Stata software (Stata Corp LP, College Station, Texas, USA); repeated measurements ANOVA using GraphPad Prism 5 (GraphPad Software; San Diego, CA, USA). A P-value less than 0.05 was considered statistically significant.
Results
All animals (n = 10) completed the experimental traumatic hemorrhage model procedure, except one animal which died (rabbit 5) at 45 min after traumatic injury, for which the end samples were taken immediately at the point of death. Mean arterial pressure (MAP) was kept around 10 mmHg during the hemorrhage phase and the end-mean value at 75 min was 9.5 ± 2.1 mmHg (). The body temperature decreased from 36.2 ± 1.3°C at baseline to 29.4 ± 1.1°C at 75 min after hemorrhage (P < .001).
Table I. Laboratory parameters, blood chemistry, and coagulation status, at baseline and 75 min after trauma with 40% hemorrhage. Data are presented as mean and standard deviation. P < .05 was considered statistically significant.
Hematological Laboratory Parameters
The physiological responses after traumatic hemorrhage included a rise in lactate from 1.0 ± 0.44 mM to 14.2 ± 3.1 mM (P < .001), a decrease in base excess from 4.2 ± 2.4 mEq/L to −15.8 ± 3.8 mEq/L (P < .001), and a dropping tendency of pH from 7.45 ± 0.005 to 7.33 ± 0.2 (P = .11) (). Blood calcium levels were largely unchanged, from a baseline value of 1.58 ± 0.06 to 1.62 ± 0.09 mM (P = .3).
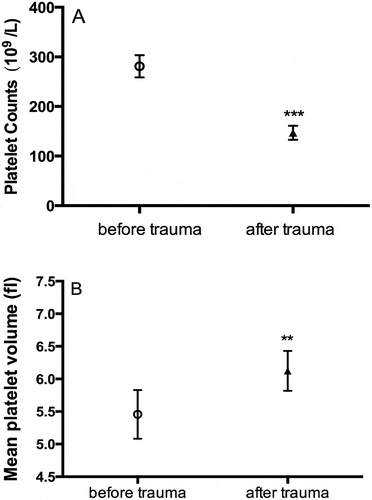
Platelet count decreased from a mean of 244 ± 80.5 x 109/L to 94.1 ± 58.5 x 109/L (P = .005), but mean platelet volume (MPV) increased from 5.4 ± 0.1 fl to 6.1 ± 0.1 fl (P = .002) (). Changes of platelet count as measured by the blood cell analyzer ABX Micros 60 were further confirmed by flow cytometric analyses of platelet event rate reduction (61.5 ± 13.7% by traumatic injury, P < .01). Hemoglobin levels decreased from 132 ± 3.1 g/L to 54 ± 6.6 g/L (P = .008) and fibrinogen levels dropped non-significantly from 1.7 ± 0.05 mg/L to 1.2 ± 0.6 mg/L (P = .453).
Figure 2. Trauma hemorrhage decreases platelet counts, but increases mean platelet volume. Platelet counts (panel A) and mean platelet volumes (panel B) were analyzed. Data plotted are mean±SEM. **P < .01, ***P < .001; n = 10.
Rotational Thromboelastometry
ROTEM® Fibtem A5 (clot firmness after 5 min) tended to decrease from 7.6 ± 3.5 mm to 5.1 ± 1.7 mm (P = .16). The same tendency was measured in ROTEM® Extem A5 with a non-significant decrease from 36.1 ± 17.5 mm to 26.4 ± 4.7 mm (P = .15) and in ROTEM® Fibtem MCF from 10.1 ± 5.8 mm to 5.8 ± 0.1 mm (P = .11). A non-significant increase was observed in clotting time for ROTEM® Intem CT from 208.1 ± 65.7 s to 276 ± 34.1 s (P = .06) and no change in ROTEM® Extem CT (50.3 s to 49.8 s; P = .87). ROTEM® Platelet ADP (AUC) tended to fall from a baseline value of 15.6 ± 10.9 Ohms*minutes to 7.8 ± 4.5 Ohms*minutes (P = .07) 75 min after trauma.
Impedance Aggregometry
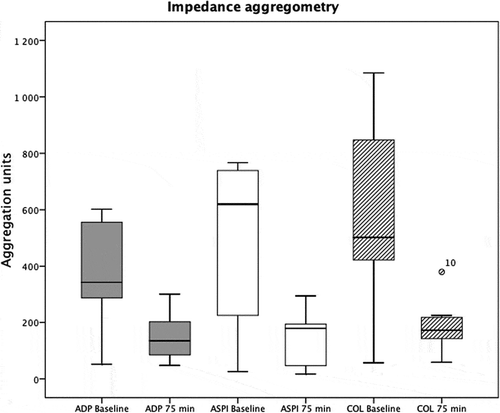
A decreased platelet aggregation response was observed for all of the studied agonists. For ADP stimulation the aggregation decreased from 376 ± 189 u to 152 ± 87 u (P = .008) and aggregation from collagen stimulation decreased from 571 ± 341 u to 178 ± 91 u (P = .007). Also, using the ASPI test, aggregation decreased from 473 ± 82 u to 139 ± 89 u (P = .021) ().
Figure 3. Trauma hemorrhage reduces platelet aggregation as assessed by impedance aggregometry. Platelet agonists were adenosine diphosphate (ADP), arachidonic acid (ASPI), and collagen (COL).
Platelet Activation as Assessed by Fibrinogen Binding
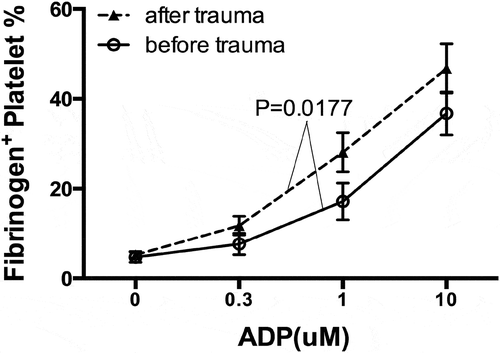
shows that ADP stimulation-induced dose-dependent platelet activation. Thus, platelet fibrinogen binding of unstimulated platelets (4.7 ± 1.1%) was elevated to 17.2 ± 4.1% by 1 µM ADP and further to 36.8 ± 4.8% by 10 µM ADP. ADP-induced platelet fibrinogen binding was enhanced after trauma (P = .0177). For example, 10 µM ADP provoked a surge of platelet fibrinogen binding to 46.8 ± 5.5% in the post-trauma samples (P < .05).
Figure 4. Trauma induces platelet hyperreactivity. Platelet fibrinogen binding was analyzed by whole blood flow cytometry using FITC-conjugated anti-fibrinogen polyclonal antibody in the absence or presence of adenosine diphosphate (ADP) stimulation. Data plotted as mean±SEM; n = 10.
Discussion
The present study used a rabbit model of experimental traumatic hemorrhage to evaluate early platelet responses to severe injury. The physiological response to traumatic injury and shock was confirmed by acidosis from the blood gases as well as a loss of temperature and blood pressure. The injury resulted in a coagulopathy, seen as a longer clotting time and a weaker blood clot by rotational thromboelastometry. After traumatic hemorrhage, the platelet count decreased, whilst the mean platelet volume increased. Platelet aggregation was significantly decreased as measured with impedance aggregometry and specific platelet rotational thromboelastometry. However, shown by flow cytometry, platelet fibrinogen binding evoked by ADP stimulation was increased after the injury.
The present model of experimental multi-trauma mimicked severe hemorrhagic shock with a consistent physiologic and metabolic response. Hypothermia, acidosis, and dilution are classically considered to be the three initiators for traumatic coagulopathy [Citation3,Citation4]. Acidosis correlates with high INR, inhibits thrombin generation, and increases fibrinogen degradation [Citation4,Citation20]. Although not statistically significant, decreased fibrinogen and increased INR indicated the development of TIC in this model. All animals also reached ROTEM® Extem A5 below 35 mm after traumatic hemorrhage but the response varied depending on baseline values. From retrospective human studies, TIC has been defined as a laboratory prothrombin time ratio >1.2 [Citation8,Citation21]. A rotational thromboelastometry clot amplitude at a 5-min threshold of <35 mm has shown a detection rate of 77% for acute traumatic coagulopathy with a false-positive rate of 13% [Citation21]. However, no consensus statement regarding the clinical presentation of TIC exists and there are no prospectively validated diagnostic criteria [Citation22,Citation23]. It may be problematic to translate human TIC criteria to animal models and we decided not to claim that all these criteria were fulfilled in this experimental model.
Blood loss resulted in a marked decrease in platelet count. The mean platelet volume was, however, markedly increased, which suggests an enhanced and compensatory platelet release from the bone marrow and the spleen after trauma. In a nonhuman primate model, platelet count decreased over time after trauma, but no animals developed thrombocytopenia with platelet count <100x103/μL.14 In a rat model of hemorrhagic shock, hypocoagulopathy appeared to involve a two-step process of platelet dysfunction followed by fibrinogen impairment [Citation24]. Post-trauma platelet counts may have a major clinical impact. Solomon et al. reported that, among patients admitted to the emergency room after sustaining a traumatic injury, the median platelet counts were lower among non-survivors than survivors (181,000/μl vs. 212,000/μl). Moreover, even mild decreases in platelet function upon admission were a sign of coagulopathy and were associated with an increased mortality [Citation9]. There are many possible factors that may contribute to platelet dysfunction after traumatic bleeding. Platelet intrinsic factors contribute to the platelet heterogeneity including size and volume but also age, levels of membrane receptors, and protein expression profile. [Citation25] Recently, Ting et al. interestingly measured the contractile force of platelet aggregates under microfluidic shear gradients for the detection of platelet dysfunction and monitoring traumatic bleeding risk. [Citation26]
Platelet functional analyses in the present study showed apparently contradicting results. Platelet aggregation was reduced as measured by impedance aggregometry upon stimulation by collagen, ADP, and ASPI. In contrast, platelet reactivity, as measured by ADP-induced platelet fibrinogen binding on singular platelets using flow cytometry, was markedly enhanced after trauma hemorrhage. Indeed, opposite platelet functional alterations have been reported using different methodologies. Thus, flow cytometric measurement of post-trauma platelet reactivity has been shown to have a higher level of reactivity compared to controls up to 72 h after trauma with higher levels of activation, especially during the early hours after trauma[Citation20]. There was also evidence showing an initial enhancement of platelet function when measured with a platelet function analyzer (PFA-100). [Citation27] When platelet reactivity was assessed by impedance aggregometry, Kutcher et al. reported that nearly 50% of 101 patients with critical injury and trauma had below-normal platelet response to at least one agonist at admission and that about 90% of the patients had platelet hypofunction at some time during their intensive care unit stay [Citation12]. Windeløv et al. found, however, no significant association between platelet aggregation response and injury severity score [Citation28]. Therefore, caution should be taken when interpreting the findings of post-trauma platelet function as assessed by different methodologies.
Platelet impedance aggregometry monitors the increase of impedance between the electrodes due to platelet adhesion, for which platelet concentration in the blood samples is critical. Thus, the results of reduced platelet aggregation in the present study and from the literature [Citation12] reflect the reduction of platelet count in the blood samples, i.e., indicating marked platelet consumption caused by trauma hemorrhage. In contrast, platelet reactivity measured by flow cytometry depicts platelet function on single-cell level. Our finding of post-trauma single platelet hyperreactivity was accompanied by a significant increase of the mean platelet volume, which indicate platelet releases from their storage pool of the bone marrow and the spleen and a higher proportion of young (newly released) platelets in the pool of circulating platelets [Citation29]. Moreover, the newly released platelets are known to be associated with platelet hyperreactivity [Citation23]. Together, our data suggest that trauma-induced rapid and significant platelet consumption due to the blood loss and hemostatic/thrombotic processes at the site of trauma and that the trauma may provoke the compensatory release of platelets from the bone marrow and the spleen. Therefore, these two closely linked processes result in a compensatory platelet hyperreactivity due to exaggerated platelet mobilization from the storage pool, but can easily get exhausted because of the low platelet counts. The present data are in line with an earlier report showing that major surgical stress during coronary artery bypass surgery can induce thrombocytopenia, but may also provoke platelet hyperreactivity [Citation30].
Our platelet results correlate well with other research groups studying the more commonly used point-of-care platelet function tests [Citation9,Citation14,Citation31]. Although the impedance aggregometry methods give mixed results, our results from flow cytometry may suggest another mechanism with compensatory platelet reactivity. The platelet count was decreased but not to the same extent as the hematocrit. This might be explained by a high level of pooling of platelets from the bone marrow but still a diluted blood sample might affect the majority of platelet function tests.
This study was performed using a rabbit model of trauma hemorrhage with a limited number of animals. It has, therefore, limitations when it comes to statistical power and generalizability. However, we believe that this model may well simulate the real situation with a blunt trauma mechanism, hemorrhage, and hypothermia. The model uses a timeframe similar to the real clinical trauma situation; from the time of injury, prehospital care, and finally early hospital care. The results from the animal model were reproducible and consistent throughout the experiments. Studies have supported the usefulnessof rabbits as an animal species for platelet research. Humans and rabbits have similar platelet counts and fibrinogen levels [Citation32,Citation33]. The fact that the abdominal aorta was catheterized may have limited blood flow circulating to the injured femurs, which might have decreased the effect of the fractures on coagulopathy. The hemorrhage is simulated by drawing blood from a central catheter which probably may further limit the effect of tissue damage on the coagulation. This model uses crystalloids for prehospital resuscitation, resulting in a dilution of the circulating blood, which further enhances coagulopathy. Today, blood products are often given early in severe hemorrhage but limited crystalloids, especially in the prehospital setting, are still common in most countries during the early resuscitation phase.
In conclusion, traumatic hemorrhage decreases platelet counts and platelet aggregation. On the other hand, the injury may enhance platelet mobilization from its storage pool, with larger and more reactive platelets. This dual pathophysiological response of platelets warrants further investigation. The study shows the feasibility of the present experimental traumatic hemorrhage model to study the platelet response to injury and bleeding. Further studies, both experimental and human are needed to characterize different aspects of platelet function in severe traumatic hemorrhage.
Declaration of interest
The authors report no conflicts of interest.
Additional information
Funding
References
- Sauaia A, Moore FA, Moore EE, Moser KS, Brennan R, Read RA, Pons PT. Epidemiology of trauma deaths: a reassessment. J Trauma 1995;38:185–193. doi:10.1097/00005373-199502000-00006
- Peden MM, McGee K, Krug E & World Health Organization. Injuries and Violence Prevention Department. Injury: a leading cause of the global burden of disease, 2000. Dept. of Injuries and Violence Prevention, Noncommunicable Diseases and Mental Health Cluster, World Health Organization, Geneva; 2002.
- Maegele M, Paffrath T, Bouillon B. Acute traumatic coagulopathy in severe injury: incidence, risk stratification, and treatment options. Dtsch Arztebl Int 2011;108:827–835. doi:10.3238/arztebl.2011.0827
- Kutcher ME, Howard BM, Sperry JL, Hubbard AE, Decker AL, Cuschieri J, Minei JP, Moore EE, Brownstein BH, Maier RV, et al. Evolving beyond the vicious triad: differential mediation of traumatic coagulopathy by injury, shock, and resuscitation. J Trauma Acute Care Surg 2015;78:516–523. doi:10.1097/TA.0000000000000545
- Maegele M, Lefering R, Yucel N, Tjardes T, Rixen D, Paffrath T, Simanski C, Neugebauer E, Bouillon B. Early coagulopathy in multiple injury: an analysis from the German Trauma Registry on 8724 patients. Injury 2007;38:298–304. doi:10.1016/j.injury.2006.10.003
- Davenport RA, Brohi K. Cause of trauma-induced coagulopathy. Curr Opin Anaesthesiol 2016;29:212–219. doi:10.1097/ACO.0000000000000295
- Brown LM, Call MS, Margaret Knudson M, Cohen MJ, Holcomb JB, Wade CE, Brasel KJ, Vercruysse G, MacLeod J, Dutton RP, et al. A normal platelet count may not be enough: the impact of admission platelet count on mortality and transfusion in severely injured trauma patients. J Trauma 2011;71:S337–342. doi:10.1097/TA.0b013e318227f67c
- Davenport RA, Brohi K. Coagulopathy in trauma patients: importance of thrombocyte function?. Curr Opin Anaesthesiol 2009;22:261–266. doi:10.1097/ACO.0b013e328325a6d9
- Solomon C, Traintinger S, Ziegler B, Hanke A, Rahe-Meyer N, Voelckel W, Schöchl H. Platelet function following trauma. A multiple electrode aggregometry study. Thromb Haemost 2011;106:322–330. doi:10.1160/TH11-03-0175
- Ramsey MT, Fabian TC, Shahan CP, Sharpe JP, Mabry SE, Weinberg JA, Croce MA, Jennings LK. A prospective study of platelet function in trauma patients. J Trauma Acute Care Surg 2016;80:726–732; discussion 732–723. doi:10.1097/TA.0000000000001017
- MacLeod JB, Lynn M, McKenney MG, Cohn SM, Murtha M. Early coagulopathy predicts mortality in trauma. J Trauma 2003;55:39–44. doi:10.1097/01.TA.0000075338.21177.EF
- Kutcher ME, Redick BJ, McCreery RC, Crane IM, Greenberg MD, Cachola LM, Nelson MF, Cohen MJ. Characterization of platelet dysfunction after trauma. J Trauma Acute Care Surg 2012;73:13–19. doi:10.1097/TA.0b013e318256deab
- Wohlauer MV, Moore EE, Thomas S, Sauaia A, Evans E, Harr J, Silliman CC, Ploplis V, Castellino FJ, Walsh M. Early platelet dysfunction: an unrecognized role in the acute coagulopathy of trauma. J Am Coll Surg 2012;214:739–746. doi:10.1016/j.jamcollsurg.2012.01.050
- Schaub LJ, Moore HB, Cap AP, Glaser JJ, Moore EE, Sheppard FR. Nonhuman primate model of polytraumatic hemorrhagic shock recapitulates early platelet dysfunction observed following severe injury in humans. J Trauma Acute Care Surg 2017;82:461–469. doi:10.1097/TA.0000000000001343
- Frith D, Cohen MJ, Brohi K. Animal models of trauma-induced coagulopathy. Thromb Res 2012;129:551–556. doi:10.1016/j.thromres.2011.11.053
- Larsson M, Forsman P, Hedenqvist P, Östlund A, Hultman J, Wikman A, Riddez L, Frenckner B, Bottai M, Wahlgren CM. Extracorporeal membrane oxygenation improves coagulopathy in an experimental traumatic hemorrhagic model. Eur J Trauma Emerg Surg. 2017;43:701-709. doi: 10.1007/s00068-016-0730-1.
- Li N, Hu H, Hjemdahl P. Aspirin treatment does not attenuate platelet or leukocyte activation as monitored by whole blood flow cytometry. Thromb Res 2003;111:165–170. doi:10.1016/j.thromres.2003.08.026
- Rothwell SW, Maglasang P, Krishnamurti C. Survival of fresh human platelets in a rabbit model as traced by flow cytometry. Transfusion 1998;38:550–556. doi:10.1046/j.1537-2995.1998.38698326334.x
- Berglund U, Lindahl T. Enhanced onset of platelet inhibition with a loading dose of ticlopedine in ASA-treated stable coronary patients. Int J Cardiol 1998;64:215–217. doi:10.1016/S0167-5273(98)00023-0
- Cheddie S, Muckart DJ, Hardcastle TC. Base deficit as an early marker of coagulopathy in trauma. S Afr J Surg 2013;51:88–90. doi:10.7196/sajs.1665
- Davenport R, Manson J, De’Ath H, Platton S, Coates A, Allard S, Hart D, Pearse R, Pasi KJ, MacCallum P, et al. Functional definition and characterization of acute traumatic coagulopathy. Crit Care Med 2011;39:2652–2658. doi:10.1097/CCM.0b013e3182281af5
- Petros S. Trauma-Induced Coagulopathy. Hamostaseologie 2019;39:20–27. doi:10.1055/s-0039-1677853
- Duan K, Yu W, Li N. The Pathophysiology and Management of Acute Traumatic Coagulopathy. Clin Appl Thromb Hemost 2015;21:645–652. doi:10.1177/1076029613516190
- Letson HL, Dobson GP. Differential contributions of platelets and fibrinogen to early coagulopathy in a rat model of hemorrhagic shock. Thromb Res 2016;141:58–65. doi:10.1016/j.thromres.2016.03.007
- van der Meijden PEJ, Heemskerk JWM. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol 2019;16:166–179. doi:10.1038/s41569-018-0110-0
- Ting LH, Feghhi S, Taparia N, Smith AO, Karchin A, Lim E, John AS, Wang X, Rue T, White NJ, et al. Contractile forces in platelet aggregates under microfluidic shear gradients reflect platelet inhibition and bleeding risk. Nat Commun 2019;10:1204. doi:10.1038/s41467-019-09150-9
- Jacoby RC, Owings JT, Holmes J, Battistella FD, Gosselin RC, Paglieroni TG. Platelet activation and function after trauma. J Trauma 2001;51:639–647. doi:10.1097/00005373-200110000-00003
- Windelov NA, Sørensen AM, Perner A, Wanscher M, Larsen CF, Ostrowski SR, Johansson PI, Rasmussen LS. Platelet aggregation following trauma: a prospective study. Blood Coagul Fibrinolysis 2014;25:67–73. doi:10.1097/MBC.0b013e328364c2da
- Spectre G, Arnetz L, Östenson C-G, Brismar K, Li N, Hjemdahl P. Twice daily dosing of aspirin improves platelet inhibition in whole blood in patients with type 2 diabetes mellitus and micro- or macrovascular complications. Thromb Haemost 2011;106:491–499. doi:10.1160/TH11-04-0216
- Li N, Astudillo R, Ivert T, Hjemdahl P. Biphasic pro-thrombotic and inflammatory responses after coronary artery bypass surgery. J Thromb Haemost 2003;1:470–476.
- Verni CC, Davila A Jr., Balian S, Sims CA, Diamond SL. Platelet dysfunction during trauma involves diverse signaling pathways and an inhibitory activity in patient-derived plasma. J Trauma Acute Care Surg 2019;86:250–259. doi:10.1097/TA.0000000000002140
- Siller-Matula JM, Plasenzotti R, Spiel A, Quehenberger P, Jilma B. Interspecies differences in coagulation profile. Thromb Haemost 2008;100:397–404.
- Hoefer IE, Grundmann S, Schirmer S, van Royen N, Meder B, Bode C, Piek JJ, Buschmann IR. Aspirin, but not clopidogrel, reduces collateral conductance in a rabbit model of femoral artery occlusion. J Am Coll Cardiol 2005;46:994–1001. doi:10.1016/j.jacc.2005.02.094