
Abstract
Platelet lifespan is regulated by intrinsic apoptosis. Platelet apoptosis can be triggered by BH3 mimetics that inhibit the pro-survival Bcl-2 family protein, Bcl-xL. Here, we investigated several small molecules that are reported to act as BH3 mimetics and compared their effects to the well-established BH3 mimetic, ABT-737. Platelet phosphatidylserine (PS) exposure was determined by flow cytometry. Changes in cytosolic Ca2+ signaling were detected using Cal-520. Plasma membrane integrity was determined by calcein leakage. The roles of caspases and calpain in these processes were determined using Q-VD-OPh and calpeptin, respectively. As previously reported, ABT-737 triggered PS exposure in a caspase-dependent manner and calcein loss in a caspase and calpain-dependent manner. In contrast, AT-101 and sabutoclax triggered PS exposure independently of caspases. HA14-1 also triggered PS exposure in a caspase-independent but calpain-dependent manner. There were also significant differences in the pattern and protease-dependency of cytosolic Ca2+ signaling in response to these drugs compared to ABT-737. Since there are clear differences between the action of ABT-737 and the other putative BH3 mimetics investigated here, AT-101, HA14-1 and sabutoclax cannot be considered as acting as BH3 mimetics in platelets. Furthermore, the platelet death caused by these drugs is likely to be distinct from apoptosis.
Introduction
Platelet lifespan is regulated by the intrinsic apoptotic pathway. Platelet apoptosis can be triggered by BH3 mimetics that inhibit the pro-survival Bcl-2 family protein, Bcl-xL [Citation1,Citation2]. Such BH3 mimetics have become useful tools to understand the mechanisms that regulate platelet apoptosis. ABT-737, the BH3 mimetic most well-characterized in platelets, inhibits Bcl-2 and Bcl-xL, triggering caspase activation, phosphatidylserine (PS) exposure in a Bak/Bax-dependent manner[Citation3], followed by release of PS-exposing extracellular vesicles (EVs) and secondary necrosis in vitro. [Citation4] The orally available analogue, ABT-263 (navitoclax), also triggers platelet apoptosis and is associated with dose-limiting thrombocytopenia[Citation5]. In contrast, ABT-199 (venetoclax), which is more selective for Bcl-2 over Bcl-xL, spares platelets and has been approved for treatment of chronic lymphocytic leukemia [Citation6,Citation7]. It is therefore important to understand which drugs trigger apoptosis in platelets.
Table I. Summary of pharmacology of putative BH3 mimetics. The effect of QVD-Oph (Q), calpeptin (C), R5421 (R) or lack of extracellular CaCl2 (No Ca) on the responses measured is shown. ‘x’ indicates that the treatment inhibited the response; ‘+’ indicates that the treatment increased the response; ‘-‘ indicates that the putative BH3 mimetic did not cause that response
In this study, we investigated several small molecules that are reported to act as BH3 mimetics. AT-101, the (-) enantiomer of gossypol, inhibits Mcl-1, Bcl-2 and Bcl-xL[Citation8], and induced caspase-dependent apoptosis in various cancer cell lines both in vivo and in vitro. [Citation9,Citation10] The pan-Bcl-2 family inhibitor, sabutoclax (BI-97C1), is also structurally related to gossypol and suppressed tumor cell growth via activation of BAK and BAX both in vivo and in vitro[Citation11]. TW-37 is a relatively more selective inhibitor of Bcl-2 derived by structure-based design from AT-101[Citation8]. TW-37 inhibited proliferation of lymphoma cells without significant effect on normal peripheral blood lymphocytes[Citation12]. HA14-1, a ligand of a Bcl-2 surface pocket, showed cytotoxic effects in vitro and in vivo, either alone or in combination with cytotoxic therapy. [Citation13–15]
We compared their effects on human platelets in vitro to those caused by ABT-737. However, although we found that some trigger platelet death, the mechanism and pharmacology of platelet death does not resemble that caused by ABT-737, suggesting that they are not acting purely as BH3 mimetics.
Methods
Washed Platelet Preparation
Blood was drawn by venepuncture into sodium citrate (3.2% v/v) from healthy, drug-free volunteers, who had given written, informed consent in accordance with the Declaration of Helsinki. Use of human blood for these experiments was approved by the Human Biology Research Ethics Committee, University of Cambridge. Washed platelets were prepared at 5 × 107 platelets/ml as previously described[Citation16]. Where required, platelets were incubated for 10 minutes with calcein-acetoxymethyl (AM) ester (1 μM; 10 min) or Cal-520-AM (500 nM; 10 min). Platelets were rested (30°C, 30 minutes) prior to stimulation with pro-apoptotic drugs. CaCl2 (2 mM) was added immediately prior to stimulation.
Flow Cytometry
Following stimulation, samples were stained with fluorescein isothiocyanate (FITC)-conjugated annexin V (AnV) (eBioscience), to detect exposed PS (FL1), unless otherwise indicated, and PE-Cy7-conjugated anti-CD41 antibody (eBioscience), to distinguish platelet-derived events. Samples were analyzed using a BD Accuri C6 flow cytometer. PE-Cy7 fluorescence (FL3) was used to trigger event acquisition. PS-positive platelet-derived EVs were defined as CD41+/AnV-positive (AnV+) events that were smaller than 1 μm. The 1 μm gate was set in forward scatter using 1 μm silica beads[Citation16]. To monitor mitochondrial membrane potential, washed platelets (5 x 107/ml) were incubated with trimetylrhodamine methyl ester (TMRM; 500 nM, 30°C, 30 min; ThermoFisher, U.K.) prior to treatment as indicated in the main text. TMRM fluorescence was acquired on FL2.
Measurement of Cytosolic Ca2+ Concentration ([Ca2+]cyt)
Cal-520-loaded platelets were stimulated in black-walled microplates at 30°C. Fluorescence (excitation: 485 nm; emission: 520 nm) was recorded using a FLUOStar OMEGA (BMG LabTech).
Sources of Materials
Reagents were from Sigma (Poole, Dorset, U.K.) unless otherwise stated. ABT-263, ABT-737, ABT-199, AT-101, HA14-1, sabutoclax and TW-37 were from Selleck Chemicals (Stratech Scientific, Ely, U.K.). Cal-520 acetoxymethyl (AM) ester was from ATT Bioquest (Sunnyvale, CA, U.S.A.). Calcein AM was from Thermo Fisher Scientific (Loughborough, U.K.). R5421 was from EndoTherm (Saarbrücken, Germany).
Data Analysis
Data are reported as mean ± standard error of mean (SEM) from at least 5 independent experiments and compared by one-way ANOVA in GraphPad Prism.
Results
Comparison of the Effects of Putative BH3 Mimetics on PS Exposure and Release of PS-exposing EVs
To compare the effects of commercially-available BH3 mimetics, platelets were treated with ABT-737, ABT-263, ABT-199, HA14-1, TW-37, AT-101, and sabutoclax (0.1, 1, 10 µM) for 3 hours. This late timepoint was chosen as the BH3 mimetic most well-characterized in platelets, ABT-737, requires several hours of treatment to see the largest effects [Citation1–4,Citation17]. Platelet PS exposure was detected using annexin V (AnV) and procoagulant extracellular vesicles (EVs) were detected as CD41+, AnV+ events less than 1 µm, as previously described[Citation16].
ABT-737 increased the percentage of AnV+ platelets in a concentration-dependent manner, which was accompanied by release of AnV+ EVs (), as previously reported[Citation4]. ABT-263 had a similar effect but was less potent. ABT-199 did not increase AnV binding up to 10 μM under these conditions, consistent with ABT-737 and ABT-263 inhibiting Bcl-xL and Bcl-2, but ABT-199 being selective for Bcl-2[Citation6]. HA14-1, AT-101 and sabutoclax all triggered platelet AnV binding and AnV+ EV release at 10 µM. TW-37 had a very weak effect, with only 5.0 ± 0.9% AnV+ platelets at 10 µM. This may reflect its low affinity to Bcl-xL, reported as 1.1 µM to recombinant Bcl-xL in vitro[Citation8].
Figure 1. Effect of putative BH3 mimetics on PS exposure and PS-exposing EV release. (a) Washed human platelets were stimulated with the indicated drugs (10 µM, 3 hours), or the vehicle (DMSO, 0.1%) after which samples were stained with anti-CD41-PerCP-Cy7, and annexin V (AnV)-FITC to detect PS exposure. PerCP-Cy7 fluorescence was used to trigger acquisition of CD41+ events. The panels show density plots of events from low density (blue) to high density (red) of forward scatter (FSC) and FITC fluorescence. The vertical line separating left and right was defined by the FSC-A of 1 µm silica beads. The density plots are representative of data from 5 different donors. (b-c) The percentage of AnV +ve platelets (CD41+, > 1 μm) or AnV +ve EVs (CD41+, < 1 μm) following treatment with the indicated concentration of each drug (3 hours). Data are mean + s.e.m. (n = 5; * p < .05; *** p < .001 compared to DMSO)
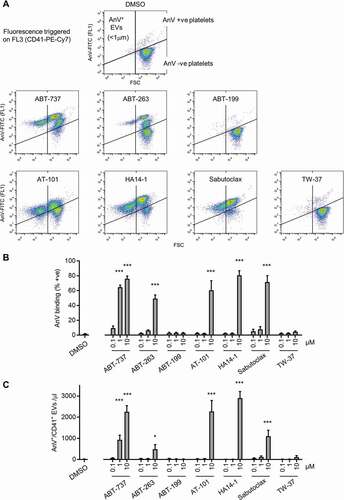
Figure 2. Timecourse of PS exposure and loss of mitochondrial membrane potential. (a) TMRM-loaded platelets were treated with the indicated drugs (10 μM). The percentage of AnV +ve platelets is shown in (a) and the percentage of TMRM +ve platelets is shown in (b). Data are mean + s.e.m. (n = 5)
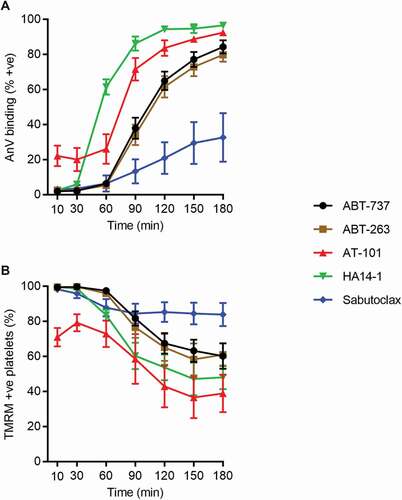
ABT-737-induced AnV binding occurred slowly (). Under our conditions, very few platelets AnV+ at 60 minutes following stimulation and 37.9 ± 6.0% platelets AnV+ by 90 minutes (Fgi. 2A). ABT-263 induced AnV binding with a similar timecourse. In contrast, HA14-1-induced AnV binding was more rapid, with 61.6 ± 4.4% platelets AnV+ after 60 minutes. AT-101-induced AnV binding showed a different pattern, with 22.1 ± 5.9% AnV+ platelets within 10 minutes. This level remained constant until 90 minutes, when it began to increase further. The effect of sabutoclax was less consistent, a feature that we found repeatably. The extent of AnV binding in response to sabutoclax varied in the 5 independent experiments (from 27.5–82.0% at 3 hours). The timecourse was similarly variable.
To investigate the effect of these putative BH3 mimetics on mitochondrial membrane potential, platelets were loaded with TMRM. This reflects late damage to the mitochondria rather than early release of cytochrome c [Citation4,Citation18]. The timecourse of loss of TMRM fluorescence matched that AnV binding. ABT-737 and ABT-263 led to a reduction in the percentage of TMRM-positive platelets. As with AnV binding, this was relatively slow. HA14-1-induced loss of TMRM fluorescence started earlier, with some platelets losing TMRM fluorescence by 60 minutes. AT-101-induced loss of TMRM fluorescence in some platelets within 10 minutes. As with AnV binding, the extent of sabutoclax–induced loss of TMRM fluorescence was variable between experiments, but largely occurred after 90 minutes.
These data suggest that ABT-263 acts in a similar manner to ABT-737, as expected. Since AT-101, HA14-1 and sabtuoclax are structurally distinct to ABT-737 yet cause PS exposure and release of PS-exposing EVs, we chose to investigate these putative BH3 mimetics in more detail.
The Putative BH3 Mimetics Show Differential Dependence on Caspase and Calpain Activity
ABT-737-induced PS exposure requires caspase activity [Citation1,Citation2]. Consistent with this, Q-VD-OPh (50 µM), a pan-caspase inhibitor, abolished AnV binding to platelets treated with ABT-737 or ABT-263 (). In contrast, Q-VD-OPh did not affect AnV+ binding in response to HA14-1, AT-101 or sabutoclax. QVD-Oph abolished AnV+ EV release in response to ABT-737, and partially inhibited AnV+ EV release in response to HA14-1 and sabutoclax ().
Figure 3. Pharmacology of PS exposure and PS-exposing EV release. Platelets were treated with Q-VD-OPh (50 µM), calpeptin (140 µM), R5421 (50 µM) or DMSO (0.1%; vehicle control) for 30 minutes prior to stimulation. CaCl2 (2 mM) was added where indicated. Platelets were then stimulated with the indicated putative BH3 mimetics (10 µM, 3 hours). The percentage of AnV +ve platelets (a) or number of AnV +ve EVs (b) was determined as in . Data are mean + s.e.m. (n = 5; * p < .05; ** p < .01; *** p < .001 compared to platelets treated with DMSO prior to stimulation with the indicated BH3 mimetic, with 2mM CaCl2)
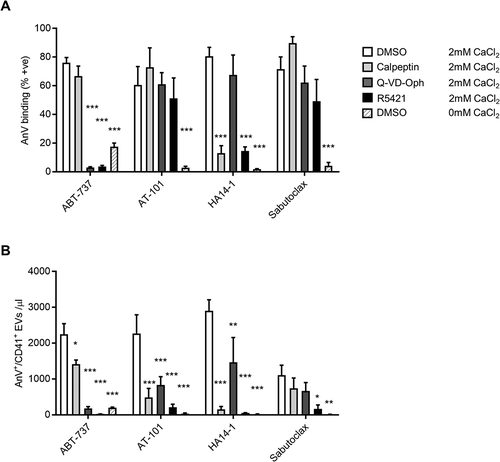
Calpeptin, a calpain inhibitor, had little effect on ABT-737, AT-101 or sabutoclax–induced AnV+ binding. In contrast, HA14-1-induced AnV+ binding was almost completely inhibited (). As previously reported, calpeptin inhibited ABT-737-induced AnV+ EV release[Citation4]. AT-101 and HA14-1 triggered AnV+ EV release to a similar level as ABT-737 and was also inhibited by calpeptinIn contrast, AnV+ EV release was lower in response to sabutoclax, and this was not significantly affected by calpeptin (Fi.g 3B).
The Putative BH3 Mimetics Show Differential Inhibition by R5421
The putative scramblase inhibitor, R5421[Citation19], also showed differential inhibition. R5421 abolished AnV+ binding in response to ABT-737, ABT-263 and HA14-1, with no inhibition of AnV+ binding in response to AT-101 or sabutoclax (). R5421 prevented AnV+ EV release in response to all putative BH3 mimetics ().
The BH3 Mimetics Show Different Effects on [Ca2+]cyt
ABT-737 treatment leads to disruption of platelet Ca2+ homeostasis [Citation1,Citation20]. To compare the effect of the different BH3 mimetics on platelet [Ca2+]cyt, platelets were loaded with the Ca2+-sensitive fluorescent dye, Cal-520. Fluorescence was recorded every minute for 3 hours following addition of the BH3 mimetic and normalized to the fluorescence of platelets at t = 0 treated with DMSO (as the vehicle) instead of the drug (F/FD). An expanded view of the first 30 minutes is in SFigure 1. ABT-737 (10 μM) increased F/FD within 5 minutes to 1.31 ± 0.09 (; see SFigure 1). This was prevented by pre-treatment with Q-VD-OPh or calpeptin, or by absence of extracellular CaCl2 (100 μM EGTA added). The small increase in fluorescence remained stable for around 60 minutes before it began to steadily increase, reaching F/FD = 3.62 ± 0.13 (n = 5) by 180 minutes. This was also inhibited by Q-VD-OPh. Calpeptin and absence of extracellular CaCl2 both partially inhibited this increased fluorescence ().
Figure 4. Effect of putative BH3 mimetics on cytosolic Ca2+ concentration. Cal-520-loaded platelets were treated with Q-VD-OPh, calpeptin, or DMSO as control prior to stimulation with the indicated drugs. 2mM CaCl2 was added (expect where EGTA, 100 µM was added instead). Fluorescence was recorded every minute for 3 hours following addition of the BH3 mimetic and normalized to the fluorescence of platelets at t = 0 treated with DMSO instead of the putative BH3 mimetic (F/FD). Traces show mean ± s.e.m. (n = 5). The mean F/FD at 5 min and 180 min is shown in (b) and (c), respectively. Data are mean + s.e.m. (n = 5; * p < .05; ** p < .01; *** p < .001 compared to DMSO-treated with 2mM CaCl2)
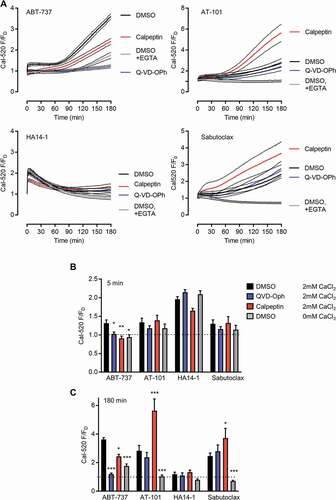
AT-101 and sabutoclax also increased Cal-520 fluorescence. Both showed a small initial increase followed by a slow but larger increase in F/FD (). The extent of the later increase was always lower than that induced by ABT-737 in matched samples. Addition of EGTA completely abolished the late increase induced by either AT-101 or sabutoclax, whereas Q-VD-OPh had little effect. Interestingly, calpeptin enhanced the late increase in [Ca2+]cyt. These data suggest that the effects of AT-101 and sabutoclax on [Ca2+]cyt are mechanistically different to the effects of ABT-737.
HA14-1, in contrast, rapidly increased F/FD to 1.96 ± 0.07 at 5 minutes. This was not significantly affected by removal of extracellular CaCl2 and addition of EGTA (F/FD = 2.11 ± 0.09 at 1 minute; n = 5; p > .05). F/FD declined from this rapid peak over the course of 2–3 hours. Q-VD-OPh had no effect on this pattern of [Ca2+]cyt signaling. Calpeptin had a small effect on the peak magnitude of the signal (). These data suggest that the effects of HA14-1 are also different to any of the other drugs tested.
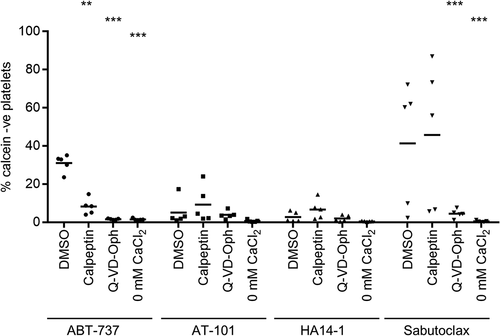
The BH3 Mimetics Differentially Affect Plasma Membrane Integrity
To assess loss of plasma membrane integrity, platelets were loaded with the fluorescent dye, calcein. Prolonged treatment with ABT-737 leads to loss of calcein fluorescence from some platelets suggesting progression from apoptosis into secondary necrosis[Citation4]. We readily repeated this observation, and the finding that calcein loss is prevented by inhibition of caspase or calpain activity (). Few platelets lost calcein fluorescence in response to AT-101 or HA14-1 treatment (5.1 ± 3.1% and 2.8 ± 1.2%, respectively), despite inducing PS exposure in a similar percentage of platelets to ABT-737 (). Sabutoclax, by contrast, induced calcein loss in 41.3 ± 14.6% (n = 5) of platelets; on average, this was unaffected by calpeptin (45.8 ± 16.9%; n = 5; p > .05) but inhibited by QVD-Oph (4.6 ± 1.0%; n = 5; p < .001) and the absence of extracellular CaCl2 (0.5 ± 0.2%; n = 5; p < .001). However, it was also notable that the effect of sabutoclax was highly variable in the 5 independent experiments, making is more difficult to draw firm conclusions regarding sabutoclax (range: 2.3–72.2%; for comparison, the range of calcein negative platelets ABT-737 in experiments paired with those with sabutoclax was 23.6–35.2%).
Figure 5. Effect of putative BH3 mimetics on plasma membrane integrity. Calcein-loaded platelets were treated with Q-VD-OPh, calpeptin, or DMSO as control prior to stimulation with the indicated drugs. 2mM CaCl2 was added except where indicated (these 0 mM CaCl2 samples also had DMSO). The percentage of calcein negative (-ve) platelets was determined by flow cytometry. Individual data (n = 5) are shown to emphasize the variability in response to sabutoclax (see main text). The horizontal bar indicates the mean. ** p < .01; *** p < .001 compared to DMSO-treated with 2mM CaCl2.
Discussion
Cancer cells often avoid apoptosis by upregulating anti-apoptotic Bcl-2 family proteins, including Bcl-2 and Bcl-xL. These anti-apoptotic proteins are antagonized by pro-apoptotic BH3-only proteins. This has made small molecule BH3 mimetics an attractive approach to trigger apoptosis in these cancer cells [Citation21,Citation22]. However, Bcl-xL is also an essential regulator of platelet lifespan[Citation3]. ABT-263 and ABT-737, which inhibit Bcl-xL, trigger the intrinsic apoptosis pathway in platelets, leading to PS exposure in vitro and thrombocytopenia in vivo [Citation1,Citation2].
ABT-737, −263 and −199 are not the only molecules reported to act as BH3 mimetics. Several pro-apoptotic drugs that have been reported to be BH3 mimetics are molecules related to gossypol, a natural phenol found in cotton plants, and which may underlie the toxicity of cotton seed[Citation23]. These include AT-101, the (-) enantiomer of gossypol, sabutoclax, and TW-37. In addition, HA14-1 is a structurally distinct small molecule that also reportedly acts as a BH3 mimetic. We are not aware of any studies investigating whether these drugs cause thrombocytopenia in patients or animal models in vivo.
As expected, ABT-737 and ABT-263 triggered PS exposure in a concentration-dependent manner, whereas ABT-199 had no effect. TW-37 also had little effect on platelets. TW-37 was developed by structure-based design from AT-101 to be more selective for Bcl-2 over Bcl-xL[Citation8], in an analogous manner to the later development of ABT-199 from ABT-263. The lack of effect of TW-37 and ABT-199 is consistent with the lack of role of Bcl-2 in mouse platelet survival[Citation24]. AT-101, HA14-1 and sabutoclax also triggered PS exposure and release of PS-exposing EVs. Since AT-101, HA14-1 and sabtuoclax are structurally distinct to ABT-737 yet cause PS exposure and release of PS-exposing EVs, we chose to investigate these putative BH3 mimetics in more detail.
The pharmacology of platelet PS exposure indicates that these molecules are acting through different mechanisms (summarised in ). PS exposure triggered by ABT-737 was dependent on caspases, as it was abolished by a pan-caspase inhibitor. In contrast, this inhibitor did not affect PS exposure triggered by AT-101, HA14-1 or sabutoclax. Caspase-dependency is not per se a sufficient indictor of apoptosis[Citation25]. However, the difference between ABT-737 – whose action has been shown to be apoptosis through its dependence on Bak/Bax – and these other putative BH3 mimetics suggests that the latter act through a non-apoptotic mechanism.
The putative scramblase inhibitor, R5421, inhibited platelet PS exposure in response to ABT-737, ABT-263 and HA14-1, but had little effect on PS exposure in response to AT101 or sabutoclax. The specificity of R5421 is unclear. Previous studies demonstrate that although the Ca2+dependent scramblase, TMEM16F, partially contributes to ABT-737-induced PS exposure, some PS exposure is TMEM16F-independent. R5421 is likely to inhibit targets in addition to TMEM16F (Sarah Millington-Burgess, personal communication; see OC27.4 in ref. [Citation26]). However, whatever its targets, the differential inhibition of platelet AnV+ binding by R5421 further indicates that there are important differences in the mechanisms of action of these putative BH3 mimetics.
The pharmacology of PS-exposing EV release is also different between the putative BH3 mimetics. Interestingly, it is also different from the pharmacology of platelet PS exposure. Caspase inhibition partially inhibited release of PS-exposing EVs in response to AT-101 and HA14-1 despite not affecting platelet PS exposure. PS-exposing EV release in response to AT-101 and sabutoclax was inhibited by R5421 despite no effect on platelet PS exposure. These observations show that inhibition of PS-exposing EV release by R54421 is not simply a consequence of inhibition of PS exposure, further suggesting its lack of specificity. Moreover, the different sensitivity to inhibition compared to ABT-737 further shows that these putative BH3 mimetics have different mechanisms of action.
ABT-737 treatment disrupted intracellular Ca2+ homeostasis in a biphasic manner. There was a small initial increase in [Ca2+]cyt followed by a later and larger increase. The small initial increase may be related to the small, transient increase that we have previously reported[Citation20]. The later, larger, sustained increase in [Ca2+]cyt was largely dependent on Ca2+ entry from the extracellular medium, as it was prevented by chelating extracellular Ca2+. A sustained increase in [Ca2+]i has been previously reported[Citation27]. There also appears to be a component of Ca2+ release from intracellular Ca2+stores, consistent with the depletion of these stores that has been previously reported. [Citation1,Citation20] Ca2+ entry may occur through Ca2+ channels, though some may also be passive Ca2+ entry as the platelets gradually lose plasma membrane integrity. This could account for the inhibitory effect of calpeptin, since calpeptin also inhibited the loss of plasma membrane integrity during secondary necrosis[Citation4]. Whatever the precise mechanisms by which intracellular Ca2+ homeostasis is lost, caspase activation is a key step. Caspase inhibition prevented the initial and late increases in [Ca2+]cyt.
The differences between the various BH3 mimetics are clearly seen in the different patterns of [Ca2+]cyt that they induce, and the different effects of protease inhibitors. The [Ca2+]cyt signal in response to AT-101 or sabutoclax was unaffected by Q-VD-OPh, in a similar manner to PS exposure. Interestingly, calpeptin enhanced the [Ca2+]cyt signal induced by these two BH3 mimetics. Although much more work is required to understand how this may occur, it clearly shows that the mechanisms that underlie [Ca2+]cyt signaling in response to these drugs is different to the mechanisms triggered by ABT-737.
The [Ca2+]cyt signals triggered by HA14-1 are different again. Here, they appear similar to those triggered by receptor activation, with a rapid initial peak followed by a slow decline back to baseline. This signal was largely unaffected by either Q-VD-OPh or by calpeptin. It was also unaffected by chelation of extracellular Ca2+, suggesting that it involves rapid, transient release from intracellular Ca2+ stores. In future studies it would be interesting to determine whether HA14-1 acts directly on the intracellular Ca2+ stores or acts as a receptor agonist in an off-target manner.
We have previously shown that prolonged treatment with ABT-737 in vitro leads to secondary necrosis, shown by loss of plasma membrane integrity[Citation4]. We did not observe a similar response with HA14-1 or AT-101 at 3 hours. Whether this reflects a difference in mechanism of action of these drugs, or simply a slower action than ABT-737, is not possible to determine from these data. Interestingly, the effect of sabutoclax was highly variable with a large range in the percentage of platelets that lost plasma membrane integrity. The reasons for this are unclear but suggest that any in vivo effect of sabutoclax could be similarly variable between individuals.
ABT-737 is, to our knowledge, the only member of this drug class whose action has been shown to depend on Bak/Bax in mouse platelets[Citation3], making it the only drug that can be clearly classified as acting as BH3 mimetics in platelets [Citation28,Citation29], although it is likely that ABT-263 acts in a similar manner. Since there are clear differences between the action of ABT-737 and the other putative BH3 mimetics investigated here, AT-101, HA14-1 and sabutoclax cannot be considered as acting as BH3 mimetics in platelets. Furthermore, the platelet death caused by these drugs is likely to be distinct from apoptosis. Studies in other cells have shown that these molecules cause apoptosis-independent cell death. Sabutoclax induces mitochondrial fragmentation[Citation30]. Sabutoclax, gossypol and apogossypol-induced cell death is independent of Bak/Bax and may also involve mitochondrial damage [Citation28,Citation31]. Although HA14-1-induced cell death is Bax-dependent in some models, it may also directly affect mitochondria and the endoplasmic reticulum[Citation32]. Although our study also argues against the use of these drugs as BH3 mimetics in platelets, it does also hint at the possibility of other cell death pathways in platelets. For example, platelet death through reactive oxygen species has been implicated for several chemotherapeutic drugs [Citation33–37]. Drug-induced non-apoptotic platelet death may be an important factor in chemotherapy-related thrombosis or thrombocytopenia and should be considered as a possible toxicity in any future trials of these or related drugs in patients or animal models.
Author Contributions
HW performed experiments, analysed data, edited the manuscript. MTH designed experiments, wrote and edited the manuscript.
Conflict of interest
The authors have no relevant scientific or financial conflicts of interest related to this work.
Supplemental Material
Download PDF (311.8 KB)Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Vogler M, Hamali HA, Sun X-M, Bampton ETW, Dinsdale D, Snowden RT, Dyer MJS, Goodall AH, Cohen GM. BCL2/BCL-X(L) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood 2011;117:7145–7154. doi:https://doi.org/10.1182/blood-2011-03-344812.
- Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, Josefsson EC, Alwis I, Ono A, Willcox A, et al. Bcl-x(L)-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 2011;118:1663–1674. doi:https://doi.org/10.1182/blood-2011-04-347849.
- Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, et al. Programmed anuclear cell death delimits platelet life span. Cell 2007;128:1173–1186. doi:https://doi.org/10.1016/j.cell.2007.01.037.
- Wei H, Harper MT. ABT-737 triggers caspase-dependent inhibition of platelet procoagulant extracellular vesicle release during apoptosis and secondary necrosis in vitro. Thromb Haemost 2019;19:1665–1674. doi:https://doi.org/10.1055/s-0039-1693694.
- Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong H, Chiu Y-L, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol 2010;11:1149–1159. doi:https://doi.org/10.1016/S1470-2045(10)70261-8.
- Souers A, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013;19:202–208. doi:https://doi.org/10.1038/nm.3048.
- Mihalyova J, Jelinek T, Growkova K, Hrdinka M, Simicek M, Hajek R. Venetoclax: A new wave in hematooncology. Exp Haematol 2018;61:10–25. doi:https://doi.org/10.1016/j.exphem.2018.02.002.
- Wang G, Nikolovska-Coleska Z, Yang C-Y, Wang R, Tang G, Guo J, Shangary S, Qiu S, Gao W, Yang D, et al. Structure-based design of potent small-molecule inhibitors of Anti-Apoptotic Bcl-2 Proteins. J Med Chem 2006;49:6139–6142. doi:https://doi.org/10.1021/jm060460o.
- Benvenuto M, Mattera R, Sticca JI, Rossi P, Cipriani C, Giganti MG, Volpi A, Modesti A, Masuelli L, Bei R, et al. Effect of the BH3 Mimetic Polyphenol (–)-Gossypol (AT-101) on the in vitro and in vivo Growth of Malignant Mesothelioma. Front Pharmacol 2018;9:1269. doi:https://doi.org/10.3389/fphar.2018.01269.
- Masuelli L, Benvenuto M, Izzi V, Zago E, Mattera R, Cerbelli B, Potenza V, Fazi S, Ciuffa S, Tresoldi I, et al. In vivo and in vitro inhibition of osteosarcoma growth by the pan Bcl-2 inhibitor AT-101. Invest New Drugs 2019. doi:https://doi.org/10.1007/s10637-019-00827-y.
- Wei J, Stebbins JL, Kitada S, Dash R, Placzek W, Rega MF, Wu B, Cellitti J, Zhai D, Yang L, et al. BI-97C1, an optically pure Apogossypol derivative as pan-active inhibitor of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J Med Chem 2010;53:4166–4176. doi:https://doi.org/10.1021/jm1001265.
- Mohammad RM, Goustin AS, Aboukameel A, Chen B, Banerjee S, Wang G, Nikolovska-Coleska Z, Wang S, Al-Katib A. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin. Cancer Res 2007;13:2226–2235. doi:https://doi.org/10.1158/1078-0432.CCR-06-1574.
- Skommer J, Wlodkowic D, Mättö M, Eray M, Pelkonen J. HA14-1, a small molecule Bcl-2 antagonist, induces apoptosis and modulates action of selected anticancer drugs in follicular lymphoma B cells. Leuk Res 2006;30:322–331. doi:https://doi.org/10.1016/j.leukres.2005.08.022.
- Wang J-L, Liu D, Zhang Z-J, Shan S, Han X, Srinivasula SM, Croce CM, Alnemri ES, Huang Z. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci 2000;97:7124–7129. doi:https://doi.org/10.1073/pnas.97.13.7124.
- Manero F, Gautier F, Gallenne T, Cauquil N, Grée D, Cartron P-F, Geneste O, Grée R, Vallette FM, Juin P, et al. The small organic compound HA14-1 prevents Bcl-2 interaction with Bax to sensitize malignant glioma cells to induction of cell death. Cancer Res 2006;66:2757–2764. doi:https://doi.org/10.1158/0008-5472.CAN-05-2097.
- Wei H, Malcor J, Harper MT. Lipid rafts are essential for release of phosphatidylserine-exposing extracellular vesicles from platelets. Sci Rep 2018;8:9987. doi:https://doi.org/10.1038/s41598-018-28363-4.
- van Kruchten R, Mattheij NJA, Saunders C, Feijge MAH, Swieringa F, Wolfs JLN, Collins PW, Heemskerk JWM, Bevers EM. Both TMEM16F-dependent and TMEM16F-independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood 2013;121:1850–1857. doi:https://doi.org/10.1182/blood-2012-09-454314.
- Choo H-J, Kholmukhamedov A, Zhou C, Jobe S. Inner mitochondrial membrane disruption links apoptotic and agonist-initiated phosphatidylserine externalization in platelets. Arterioscler Thromb Vasc Biol 2017;37:1503–1512. doi:https://doi.org/10.1161/ATVBAHA.117.309473.
- Dekkers DWC, Comfurius P, Vuist WMJ, Billheimer JT, Dicker I, Weiss HJ, Zwaal RFA, Bevers EM. Impaired Ca2+-induced tyrosine phosphorylation and defective lipid scrambling in erythrocytes from a patient with Scott syndrome: A study using an inhibitor for scramblase that mimics the defect in Scott syndrome. Blood 1998;91:2133–2138. doi:https://doi.org/10.1182/blood.V91.6.2133.
- Harper MT, Poole AW. Bcl-x(L)-inhibitory BH3 mimetic ABT-737 depletes platelet calcium stores. Blood 2012;119:4337–4338. doi:https://doi.org/10.1182/blood-2012-02-413070.
- Delbridge A, Strasser A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ 2015;22:1071–1080. doi:https://doi.org/10.1038/cdd.2015.50.
- Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW, Strasser A. BH3-mimetic drugs: blazing the trail for new cancer medicines. Cancer Cell 2018;34:879–891. doi:https://doi.org/10.1016/j.ccell.2018.11.004.
- Eagle E. Chronic Toxicity of Gossypol. Science 1949;109:361. doi:https://doi.org/10.1126/science.109.2832.361.
- Debrincat MA, Pleines I, Lebois M, Lane RM, Holmes ML, Corbin J, Vandenberg CJ, Alexander WS, Ng AP, Strasser A, et al. BCL-2 is dispensable for thrombopoiesis and platelet survival. Cell Death Dis 2015;6:e1721–e1721. doi:https://doi.org/10.1038/cddis.2015.97.
- Kile B. The role of apoptosis in megakaryocytes and platelets. Br J Haematol 2014;165:217–226. doi:https://doi.org/10.1111/bjh.12757.
- Millington-Burgess SL, Rahman T, Harper MT. R5421 is not a selective inhibitor of platelet phosphatidylserine exposure. OC 27.4 in Nurses and Orals Abstracts. Res Pract Thromb Haemost 2019;3:1–228. doi:https://doi.org/10.1002/rth2.12227.
- Gyulkhandanyan AV, Mutlu A, Allen DJ, Freedman J, Leytin V. BH3-mimetic ABT-737 induces strong mitochondrial membrane depolarization in platelets but only weakly stimulates apoptotic morphological changes, platelet shrinkage and microparticle formation. Thromb Res 2014;133:73–79. doi:https://doi.org/10.1016/j.thromres.2013.10.041.
- Vogler M, Weber K, Dinsdale D, Schmitz I, Schulze-Osthoff K, Dyer MJS, Cohen GM. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ 2009;16:1030–1039. doi:https://doi.org/10.1038/cdd.2009.48.
- Soderquist RS, Eastman A. BCL2 Inhibitors as Anticancer Drugs: A Plethora of Misleading BH3 Mimetics. Mol Cancer Ther 2016;15:2011–2017. doi:https://doi.org/10.1158/1535-7163.MCT-16-0031.
- Varadarajan S, Butterworth M, Wei J, Pellecchia M, Dinsdale D, Cohen GM. Sabutoclax (BI97C1) and BI112D1, Putative inhibitors of MCL-1, Induce mitochondrial fragmentation either upstream of or independent of apoptosis. Neoplasia 2013;15:568–IN22. doi:https://doi.org/10.1593/neo.13230.
- Villalobos-Ortiz M, Ryan J, Mashaka TN, Opferman JT, Letai A. BH3 profiling discriminates on-target small molecule BH3 mimetics from putative mimetics. Cell Death Differ 2019;1–9. doi:https://doi.org/10.1038/s41418-019-0391-9.
- Bodur C, Basaga H. Bcl-2 Inhibitors: emerging drugs in cancer therapy. Curr Med Chem 2012;19:1804–1820. doi:https://doi.org/10.2174/092986712800099839.
- Harper MT. Auranofin, a thioredoxin reductase inhibitor, causes platelet death through calcium overload. Platelets 2019;30:98–104. doi:https://doi.org/10.1080/09537104.2017.1378809.
- KIM E-J, LIM K-M, KIM K-Y, BAE O-N, NOH J-Y, CHUNG S-M, SHIN S, YUN Y-P, CHUNG J-H. Doxorubicin-induced platelet cytotoxicity: a new contributory factor for doxorubicin-mediated thrombocytopenia. J Thromb Haemost 2009;7:1172–1183. doi:https://doi.org/10.1111/jth.2009.7.issue-7.
- Kim S-H, Lim K-M, Noh J-Y, Kim K, Kang S, Chang YK, Shin S, Chung J-H Doxorubicin-Induced Platelet Procoagulant Activities: an Important Clue for Chemotherapy-Associated Thrombosis. Toxicol Sci 2011;124:215–224. doi:https://doi.org/10.1093/toxsci/kfr222.
- Ma R, Bi Y, Kou J, Zhou J, Shi J. Enhanced procoagulant activity of platelets after chemotherapy in non-small cell lung cancer. Cancer Biol Ther 2017;18:627–634. doi:https://doi.org/10.1080/15384047.2017.1345387.
- Zhang W, Zhao L, Liu J, Du J, Wang Z, Ruan C, Dai K. Cisplatin induces platelet apoptosis through the ERK signaling pathway. Thromb Res 2012;130:81–91. doi:https://doi.org/10.1016/j.thromres.2012.02.013.