
Abstract
Platelet-derived extracellular polyphosphate (PolyP) is a major mediator of thrombosis. PolyP is a linear chain of inorganic phosphate (Pi) and is stored in platelet dense granules. Pi enters cells from the extracellular fluid through phosphate transporters and may be stored as PolyP. Here we show that high extracellular Pi concentration in vitro increases platelet PolyP content, in a manner dependent on phosphate transporters, IP6K and V-type ATPases. The increased PolyP also enhanced PolyP-dependent coagulation in platelet-rich plasma. These data suggest a mechanistic link between hyperphosphatemia, PolyP and enhanced coagulation, which may be important in pathologies such as chronic kidney disease.
Introduction
Platelet-derived extracellular polyphosphate (PolyP) is a major mediator of hemostasis, thrombosis, and vascular inflammation and is a promising therapeutic target [Citation1–3]. Platelets store PolyP in their dense granules and release it during platelet activation[Citation4]. Extracellular PolyPs enhance coagulation while also limiting fibrinolysis[Citation1]. Platelet PolyPs therefore link platelet granule secretion to enhanced coagulation.
PolyPs are polyanionic, linear chains of inorganic phosphate (Pi) found in both prokaryotic and eukaryotic cells. In bacteria, intracellular PolyP is involved in energy storage, protection from heavy metal toxicity, and as a molecular chaperone[Citation5], although its role in eukaryotes is less well understood. Intracellular PolyP may act as buffer to minimize fluctuations in cytosolic Pi concentration ([Pi]cyt). Since Pi is essential to signal transduction and energy metabolism, its levels in the cytosol must be tightly controlled. For example, endothelial cells undergo plasma membrane blebbing and release microparticles when challenged with high [Pi]ex[Citation6], and can undergo apoptosis[Citation7].
Normal plasma [Pi] fluctuates between approximately 1.0–1.45 mM (3.0–4.5 mg/dL phosphorus) in adults[Citation8]. High plasma [Pi] (hyperphosphatemia) can be caused acutely by excessive phosphate intake or chronically by insufficient renal excretion and contributes to the high cardiovascular risk of chronic kidney disease (CKD), which includes vascular calcification and abnormal coagulation[Citation9]. Hyperphosphatemia may also increase [Pi]cyt, since Pi enters cells from the extracellular fluid through phosphate transporters[Citation6]. Storing Pi monomers in PolyP is one potential mechanism to reduce toxicity during high Pi load. In this study, we therefore investigated the effect of high extracellular inorganic phosphate concentration ([Pi]ex) on platelets, and specifically whether this affects platelet PolyP levels.
Methods
Washed platelets were isolated from blood drawn from healthy drug-free volunteers with approval from the Human Biology Research Ethics Committee, University of Cambridge, as previously described[Citation10], except that the isolated platelets were initially resuspended in HEPES-buffered saline (HBS) without Pi. NaH2PO4 was then added to the following final concentrations (in mM): 0.34, which is the concentration we have previously used in our HBS; 1.0, which represents physiological serum [Pi]; and 2.5, which occurs in hyperphosphatemia[Citation8]. PolyP was separated from platelet lysates using silica spin columns[Citation11]. DNA or RNA was removed by treating samples with DNAase and RNAase. PolyP in the samples was stained with DAPI to measure PolyP levels[Citation12]. This fluorescence was significantly reduced by treatment with alkaline phosphatase, which degrades PolyP, by 66.9 ± 1.9% (n = 4). Clot turbidometry of PRP in response to Pi-loading in the presence or absence of TRAP-6 was measured as previously described[Citation13]. [Pi]ex in PRP showed variation between samples, though in none was [Pi]ex greater than 1.1 mM. Where [Pi]ex was lower than 1 mM, it was increased to 1 mM by addition of NaH2PO4. Hyperphosphatemia was mimicked by addition of NaH2PO4 to give [Pi]ex of 2.5 mM.
Results
Platelet PolyP content was significantly increased by incubating platelets in high [Pi]ex (2.5 mM) (). This effect was rapid, with a significant increase detected within 10 minutes. In contrast, the PolyP content of platelets was not significantly different between platelets incubated in low (0.34 mM) and physiological (1 mM) [Pi]ex. This suggests that platelets dynamically respond to pathological elevations in [Pi]ex by increasing their PolyP content.
Figure 1. High extracellular phosphate increases platelet PolyP and clotting in PRP
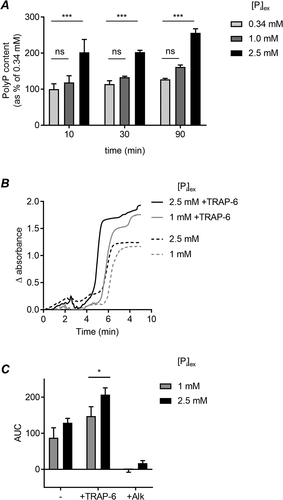
Clot turbidometry was used to investigate whether the increased PolyP content of platelets influences coagulation. Platelet-rich plasma (PRP) was prepared from blood drawn into sodium citrate (3.2%). NaH2PO4 was added to increase [Pi]ex, using the malachite green phosphate assay titrate the NaH2PO4 required to elevate [Pi]ex to 2.5 mM. After 90 minutes, coagulation was initiated by addition of CaCl2 (20 mM). Clot turbidity was measured absorbance (405 nm) in a microplate reader without shaking ( shows representative traces). The area under the curve (AUC) was measured. As expected, addition of CaCl2 triggered clotting of citrated PRP, whereas citrated PRP in the absence of additional CaCl2 did not begin to clot (data not shown). CaCl2-triggered clotting under these conditions (no exogenous tissue factor) was dependent on PolyP as it was abolished by the addition of alkaline phosphatase (). There was no significant difference in clotting at the two levels of [Pi]ex. However, if samples where platelets were stimulated with the PAR1 agonist, SFLLRN-amide, to promote platelet activation and granule secretion, the AUC was significantly increased when [Pi]ex was increased to 2.5 mM (). Together, these data suggest that high [Pi]ex promotes the procoagulant behavior of stimulated platelets by increasing PolyP levels.
To investigate whether Pi must enter platelets to increase PolyP levels, we used phosphonoformic acid (PFA; 1 mM) to block phosphate transporters. Pre-treatment with PFA prevented the increase in PolyP in response to high [Pi]ex (), indicating that Pi must enter platelets rather than act on an extracellular site.
Figure 2. Regulation of PolyP levels by high [Pi]ex.
![Figure 2. Regulation of PolyP levels by high [Pi]ex.](/cms/asset/f93cdef9-a748-46f8-9ba0-76dab11e6cde/iplt_a_1817358_f0002_b.gif)
V-type ATPases and inositol hexakisphosphate kinase 1 (IP6K1) have been previously implicated in PolyP synthesis and storage in platelets [Citation4,Citation13]. The increase in PolyP in response to high [Pi]ex was also inhibited by bafilomycin A, an inhibitor of V-type ATPases, and TNP, a potent and selective inhibitor of IP6K1 ().
Discussion
Our study shows that platelets synthesize PolyP in response to high [Pi]ex. We propose that Pi enters platelets through phosphate transporters and is converted to PolyP and stored in dense granules in a mechanism dependent on IP6K and V-type ATPases (). This mechanism may protect platelets from high [Pi]cyt load during periods of hyperphosphatemia. It also shows that PolyP levels in platelets are not fixed during platelet biogenesis but can rapidly fluctuate in response to changes in their environment. However, the stored PolyP is released when platelets are activated, increasing coagulation.
The increase in PolyP content was inhibited by PFA, which inhibits phosphate transporters. PFA also inhibits viral DNA polymerase, underpinning its clinical use as an antiviral (where it is called foscarnet). Although the concentration used in our study is high, it is possible that similar concentrations can be achieved in patients[Citation14], although blocking phosphate transporters (including in the kidney) is unlikely to be useful in hyperphosphatemia. Instead, blocking the PolyP synthesis pathway may be of more benefit.
How PolyP is synthesized and regulated in mammalian cells is not well understood. In fungi and some protists, such as Trypanosoma and Leishmania, PolyP synthesis requires a vacuolar transporter chaperone complex. PolyP that is synthesized at the cytoplasmic face of acidocalcisome-like organelles by this complex is translocated into the organelle lumen. A transmembrane proton gradient generated by V-type ATPase activity is also required[Citation15]. Notably, platelet polyphosphate is stored in dense granules, a secretory lysosome-like organelle that has an acidic lumen and is similar to acidocalcisomes[Citation4]. Since bafilomycin prevented the increase in PolyP, platelets may use the proton gradient across the dense granule membrane to synthesize and accumulate PolyP.
IP6K1 also regulates PolyP levels in eukaryotic cells. Although normal function of IP6K1 is to synthesis 5-diphosphoinositol pentakisphosphate (IP7) from inositol hexakisphosphate (IP6), Ip6k1-deficient yeast has substantially reduced levels of PolyP, suggesting a link between the metabolism of PolyP and inositol pyrophosphates. Similarly, the PolyP level in platelets from Ip6k1−/- mice is half that of wild-type platelets[Citation13]. Plasma clotting time was prolonged in the Ip6k1−/- mice. Tail bleeding time was also increased, and the mice were protected in a model of pulmonary embolism. Consistent with this, the increase in PolyP in response to high [Pi]ex was inhibited by TNP.
It would be interesting to investigate whether platelets from patients with hyperphosphatemia may have increased PolyP levels and whether this contributes to abnormal coagulation. Since it is possible to block the increase in PolyP levels, as suggested by our data, or the pro-thrombotic effects of PolyP[Citation2], PolyP is a potential pharmacological target to reduce the pathophysiology associated with hyperphosphatemia.
Acknowledgements
This work was supported by British Heart Foundation Project Grants PG/17/45/33071 and PG/16/45/32152. NA, designed the research, performed research, analyzed data, and drafted and edited the manuscript. MTH designed the research, analyzed data, and wrote the manuscript.
Disclosure Statement
The authors have no conflicts of interest relating to this work.
Additional information
Funding
References
- Travers RJ, Smith SA, Morrissey JH. Polyphosphate, platelets, and coagulation. Int J Lab Hematol 2015;37:31–35. doi:https://doi.org/10.1111/ijlh.12349
- Smith SA, Choi SH, Collins JNR, Travers RJ, Cooley BC, Morrissey JH. Inhibition of polyphosphate as a novel strategy for preventing thrombosis and inflammation. Blood 2012;120(26):5103–5110. doi:https://doi.org/10.1182/blood-2012-07-444935
- Mailer RKW, Hänel L, Allende M, Renné T. Polyphosphate as a target for interference with inflammation and thrombosis. Frontiers in Medicine 2019;6. doi:https://doi.org/10.3389/fmed.2019.00076
- Ruiz FA, Lea CR, Oldfield E, Docampo R. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J. Biol. Chem 2004;279(43):44250–44257. doi:https://doi.org/10.1074/jbc.M406261200
- Albi T, Serrano A. Inorganic polyphosphate in the microbial world. Emerging roles for a multifaceted biopolymer. World J Microbiol Biotechnol 2016;32(2):27. doi:https://doi.org/10.1007/s11274-015-1983-2
- Abbasian N, Burton JO, Herbert KE, Tregunna B-E, Brown JR, Ghaderi-Najafabadi M, Brunskill NJ, Goodall AH, Bevington A. Hyperphosphatemia, phosphoprotein phosphatases, and microparticle release in vascular endothelial cells. J Am Soc Nephrol 2015;26(9):2152–2162. doi:https://doi.org/10.1681/ASN.2014070642
- Di Marco GS, Hausberg M, Hillebrand U, Rustemeyer P, Wittkowski W, Lang D, Pavenstädt H. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am J Physiol Ren Physiol 2008;294(6):F1381–7. doi:https://doi.org/10.1152/ajprenal.00003.2008
- Goyal R, Jialal I. Hyperphosphatemia. [Updated 2020 Jun 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK551586/.
- Disthabanchong S. Vascular calcification in chronic kidney disease: pathogenesis and clinical implication. World J Nephrol 2012;1(2):43. doi:https://doi.org/10.5527/wjn.v1.i2.43
- Wei H, Davies JE, Harper MT. 2-Aminoethoxydiphenylborate (2-APB) inhibits release of phosphatidylserine-exposing extracellular vesicles from platelets. Cell Death Discov 2020;6(1):10. doi:https://doi.org/10.1038/s41420-020-0244-9
- Lee WD, Gawri R, Shiba T, Ji A-R, Stanford WL, Kandel RA. Simple silica column–based method to quantify inorganic polyphosphates in cartilage and other tissues. Cartilage 2018;9(4):417–427. doi:https://doi.org/10.1177/1947603517690856
- Schlagenhauf A, Pohl S, Haidl H, Leschnik B, Gallistl S, Muntean W. Non-enzymatic quantification of polyphosphate levels in platelet lysates and releasates. J Pharm Biomed Anal 2016;131:1–5. doi:https://doi.org/10.1016/j.jpba.2016.08.005
- Ghosh S, Shukla D, Suman K, Lakshmi BJ, Manorama R, Kumar S, Bhandari R. Inositol hexakisphosphate kinase 1 maintains hemostasis in mice by regulating platelet polyphosphate levels. Blood 2013;122(8):1478–1486. doi:https://doi.org/10.1182/blood-2013-01-481549
- Noormohamed FH, Youle MS, Higgs CJ, Martin-Munley S, Gazzard BG, Lant AF. Pharmacokinetics and absolute bioavailability of oral foscarnet in human immunodeficiency virus-seropositive patients. Antimicrob Agents Chemother 1998;42(2):293. doi:https://doi.org/10.1128/AAC.42.2.293
- Desfougères Y, Saiardi A, Azevedo C. Inorganic polyphosphate in mammals: where’s Wally? Biochem Soc Trans 2020;48(1):95–101. doi:https://doi.org/10.1042/BST20190328