
Abstract
Most agonists stimulate platelet Ca2+ rises via G-protein coupled receptors (GPCRs) or ITAM-linked receptors (ILRs). Well studied are the GPCRs stimulated by the soluble agonists thrombin (PAR1, PAR4), ADP (P2Y1, P2Y12), and thromboxane A2 (TP), signaling via phospholipase (PLC)β isoforms. The platelet ILRs glycoprotein VI (GPVI), C-type lectin-like receptor 2 (CLEC2), and FcγRIIa are stimulated by adhesive ligands or antibody complexes and signal via tyrosine protein kinases and PLCγ isoforms. Marked differences exist between the GPCR- and ILR-induced Ca2+ signaling in: (i) dependency of tyrosine phosphorylation; (ii) oscillatory versus continued Ca2+ rises by mobilization from the endoplasmic reticulum; and (iii) smaller or larger role of extracellular Ca2+ entry via STIM1/ORAI1. Co-stimulation of both types of receptors, especially by thrombin (PAR1/4) and collagen (GPVI), leads to a highly enforced Ca2+ rise, involving mitochondrial Ca2+ release, which activates the ion and phospholipid channel, anoctamin-6. This highly Ca2+-dependent process causes swelling, ballooning, and phosphatidylserine expression, establishing a unique platelet population swinging between vital and necrotic (procoagulant ‘zombie’ platelets). Additionally, the high Ca2+ status of procoagulant platelets induces a set of additional events: (i) Ca2+ dependent cleavage of signaling proteins and receptors via calpain and ADAM isoforms; (ii) microvesiculation; (iii) enhanced coagulation factor binding; and (iv) fibrin-coat formation involving transglutaminases. Given the additive roles of GPCR and ILR in Ca2+ signal generation, high-throughput screening of biomolecules or small molecules based on Ca2+ flux measurements provides a promising way to find new inhibitors interfering with prolonged high Ca2+, phosphatidylserine expression, and hence platelet procoagulant activity.
Introduction
Blood platelets respond to and become activated by multiple (patho)physiological agonists and chemically synthetized drugs. The majority of these compounds act by one out of two types of signaling pathways, i.e. via G-protein coupled receptors (GPCRs) or via ITAM-linked receptors (ILRs, immunoreceptor tyrosine-based activation motif-linked receptors) [Citation1,Citation2]. Typically, soluble agonists mostly trigger platelets via GPCRs, with as an example the coagulation product thrombin, acting on the protease-activated receptors (PAR)1 and PAR4. On the other hand, but not exclusively, vascular-bound agonists trigger platelets via ILRs, with as an example collagen, stimulating via the ILR glycoprotein VI (GPVI). Both thrombin and collagen are considered to be key agonists in (pathological) thrombus formation in man and rodents [Citation3,Citation4], and both trigger directly or indirectly a whole range of platelet responses [Citation1]. Markedly, thrombin and collagen also directly steer the coagulation process [Citation2]. Given this, it may not be surprising that also platelets have a potent regulatory role in blood clotting [Citation5], a role that is concentrated in the population of ‘procoagulant platelets’ stimulated through the membrane channel protein anoctamin-6 (previously known as TMEM16F) [Citation6].
In the present paper, we outline how platelet activation via GPCR and ILR signaling pathways, synergizing into cytosolic Ca2+ rises, induces anoctamin-6 activation, and causes procoagulant platelet formation. This also leads to a whole range of downstream responses in thrombosis and hemostasis, which culminate in the formation of a fibrin clot. Finally, we stipulate that high-throughput screening for molecules affecting the agonist-induced Ca2+ rises can provide a promising way to interfere in platelet-mediated blood clotting.
Platelet Activation via GPCRs
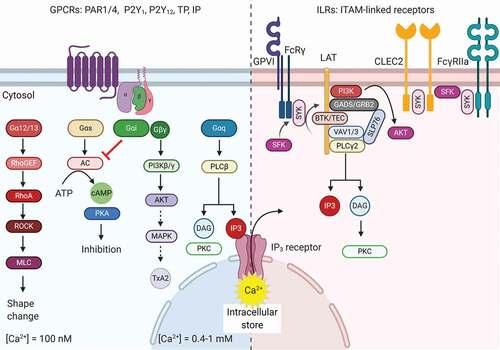
As in their name, GPCRs couple to heterotrimeric G-proteins, comprising of α, β, and γ subunits, each of them present different isoforms. For all GPCRs, receptor binding causes a switch from the inactive (GDP binding) to the active (GTP binding) form of the Gα subunits. The most relevant Gα-subunits for platelet Ca2+ signaling and ensuing processes are: (i) Gαq (raising cytosolic Ca2+ via PLCβ isoforms) and (ii) Gαs (lowering cytosolic Ca2+ via cAMP production) (). The other G-proteins, Gα12/13 (activating the monomeric GTPase, RhoA) and Gαi (suppressing cAMP production and activating phosphatidylinositol 3-kinases via βγ subunit), modulate the Ca2+ signal only indirectly, as detailed below [Citation1,Citation7]. Of the dozens of GPCRs expressed by platelets, most well studied in relation to Ca2+ signaling are the receptors for thrombin (PAR1, PAR4), ADP (P2Y1, P2Y12), and thromboxane A2 (TP). Dysfunctional mutations of most of these receptors have been described [Citation8,Citation9], as detailed below.
Figure 1. Schematic overview of the different signaling pathways in terms of calcium signaling. Left part GPCRs: G-protein coupled receptors bearing the Gαq isoform activating subunit promote phospholipase C (PLC) β to generate phospholipase inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 triggers the release of Ca2+ from intracellular stores and DAG activates several proteins, such as different isoforms of protein kinase C (PKC). Other Gα isoforms (Gα12/13, Gαs, Gαi) trigger signaling pathways that synergize or negatively regulate Ca2+ increase, and modulate integrin activation, thromboxane generation and granule release. Right part ILRs: ITAM-linked-receptor activation results in the recruitment of several tyrosine kinase proteins to LAT. The formation of this signalosome results in the activation of PLCγ2, also eliciting IP3 and DAG formation. Other abbreviations: AC: adenylyl cyclase, CLEC2: C-type lectin-like receptor 2, FcRγ: Fc receptor γ-chain, FcγRIIa: Fc γ receptor IIa, GPVI: glycoprotein VI, LAT: linker of activated T cells, MAPK: mitogen-activated protein kinase, PAR1/4: protease-activated receptor 1 and 4, PI3K: phosphatidylinositol 3-kinase, PKA: protein kinase A, PKC: protein kinase C, PLC: phospholipase C, P2Y1 and P2Y12: purinergic receptor 1 and 12, ROCK: Rho-associated protein kinase, SFK: Src family kinase, TP: thromboxane receptor, Syk: spleen tyrosine kinase, TxA2: thromboxane A2. This figure was created using BioRender.com
Protease-Activated Receptors (PARs)
Handbook knowledge is that the central coagulation protease thrombin cleaves and activates two GPCRs of human platelets, PAR1 and PAR4, both of which are directly coupled to Gαq. The isoform PAR1 is known to be more sensitive for thrombin and to prime for activation of PAR4 [Citation7,Citation10,Citation11]. PAR1 induces an acute and rather short-lived rise in Ca2+ plus initial integrin activation, whereas PAR4 causes a steady and more sustained Ca2+ signal that also depends on autocrine P2Y12-mediated events. Accordingly, in thrombin-stimulated platelets, the total amount of Ca2+ signal is the time-integrated sum of contributions of both receptors () [Citation12,Citation16,Citation17]. In mouse platelets, PAR3 as a non-cleaved receptor replaces PAR1 [Citation7]. Regardless of the species, the more prolonged PAR4 signaling is considered to be needed for the platelet procoagulant response and for thrombus stabilization [Citation18,Citation19]. Part of the PAR-dependent signaling can occur independently of Ca2+ mobilization via a Gα12/13 pathway, which contributes to the platelet shape-change response [Citation20].
Figure 2. Schematic representation of the different pattern of calcium responses in a single platelet stimulated by GPCRs or ILRs agonists. Fura2-labeled platelets were allowed to adhere to fibrinogen (A) or collagen (B, C) surfaces. Thrombin addition is indicated by an arrow. A) The GPCR agonist, thrombin, promotes oscillatory calcium traces in single platelets calcium signaling, while B) collagen, as an agonist of ILRs, promotes a sustained calcium rise. C) Procoagulant response, due to anoctamin-6 activation, was triggered by a continued high calcium rise after stimulation by collagen and thrombin. Schematic traces are based on published measurements in single platelets [Citation12–15].[Citation103]
![Figure 2. Schematic representation of the different pattern of calcium responses in a single platelet stimulated by GPCRs or ILRs agonists. Fura2-labeled platelets were allowed to adhere to fibrinogen (A) or collagen (B, C) surfaces. Thrombin addition is indicated by an arrow. A) The GPCR agonist, thrombin, promotes oscillatory calcium traces in single platelets calcium signaling, while B) collagen, as an agonist of ILRs, promotes a sustained calcium rise. C) Procoagulant response, due to anoctamin-6 activation, was triggered by a continued high calcium rise after stimulation by collagen and thrombin. Schematic traces are based on published measurements in single platelets [Citation12–15].[Citation103]](/cms/asset/bdaed714-03a2-433c-a1a0-e2119db8a52f/iplt_a_1859103_f0002_b.gif)
Several human PAR4 variants (gene F2RL3) are known to link to alterations in platelet responses [Citation8,Citation21]. Furthermore, blocking of PAR4 or its hyperreactive forms was found to provide an antithrombotic potential [Citation22,Citation23]. Regarding the other receptor PAR1 (gene F2R), antagonists like vorapaxar are being used in some countries for secondary prevention of atherothrombotic disease [Citation24]. Current judgment is that better understanding of the interindividual variation in the PARs’ activities is needed to come to a better personalized medication [Citation25].
Purinergic Receptors P2Y1 and P2Y12
The two key purinergic GPCRs for ADP in platelets are P2Y1 and P2Y12 [Citation26]. While ADP is considered to be a weak platelet agonist, it plays an important role in the amplification and stabilization of platelets responses to many other receptor agonists, including thrombin and thromboxane A2 [Citation27]. The low-level P2Y1 receptors signal via Gαq- and PLCβ-mediated Ca2+ rises, whereas the more abundant P2Y12 receptors are coupled to Gαi [Citation26]. The relatively small Ca2+ signal evoked by P2Y1 contributes to platelet shape change. On the other hand, the P2Y12 pathway involves inhibition of adenylyl cyclase activity via the Gαi subunit and activation of phosphatidylinositol 3-kinases (PI3Ks) via the βγ subunits, the latter directing to integrin αIIbβ3 activation [Citation1]. Markedly, P2Y12 was also found to contribute to the thrombin-induced exposure of phosphatidylserine (PS) in isolated platelets [Citation28].
The literature provides also ample evidence for synergic action between the P2Y1 and P2Y12 receptors. Recent work shows that in rat megakaryocytes P2Y12 signaling can enhance the P2Y1-induced Ca2+ oscillations, which then stimulates integrin αIIbβ3 activation and fibrinogen binding [Citation29]. This adds well to earlier evidence that P2Y12 contributes to platelet activation by potentiation of the P2Y1-induced Ca2+ responses both via inhibition of adenylate cyclase and activation of PI3Ks with a complex additional role of Src kinase [Citation30]. Critical in this interplay of Ca2+-dependent platelet activation via both P2Y receptors is also the guanine nucleotide exchange factor CalDAG-GEFI, which in a Ca2+-dependent way regulates integrin activation through Rap1b [Citation31].
Dysfunctional (gain- or loss-of-function) variants of the genes for both P2Y1 and P2Y12 (P2RY1, P2YR12) have been identified, which associate with a bleeding or a prothrombotic propensity [Citation32,Citation33]. As an illustration of the relevance of the P2Y12-PI3K-αIIbβ3 activation pathway, (pro)drugs inhibiting this receptor are widely used for the secondary prevention of arterial thrombosis, even as monotherapy. Mostly prescribed drugs are thienopyridines that irreversibly block the receptor (prodrugs: clopidogrel and prasugrel), and the compounds ticagrelor and cangrelor that reversibly bind to P2Y12 [Citation34].
Thromboxane A2 Receptor (TP)
The TP receptor for thromboxane A2 is expressed on platelets and on vascular cells, and it couples to Gαq (regulating Ca2+ release) and Gα12 family proteins (regulating integrin activation) [Citation7,Citation35]. Aspirin, one of the hallmarks of current anti-platelet therapy, blocks the formation of thromboxane A2, thus preventing its interaction with the TP receptor. Without this drug, the autocrine platelet-released thromboxane A2 greatly enhances the responses of other platelet agonists, in particular ADP and collagen [Citation2]. Patients with a mutation in the TBXA2R gene develop bleeding symptoms, and their platelets react less to thromboxane A2, ADP, and other agonists [Citation36,Citation37]. In the past, several efforts have been made to develop specific TP antagonists. The extra advantage of such antagonists was questioned, given the high clinical importance of aspirin as a ‘gold-standard’ anti-platelet drug for the prevention of cardiovascular events. However, it is well possible that receptor antagonists have less bleeding side effects.
Other GPCRs
In the context of Ca2+ signal generation, another relevant GPCR on platelets is the IP receptor for endothelial-derived prostacyclin. This receptor couples to Gαs and raises the cAMP level, which downregulates Ca2+ rises via protein kinase A [Citation38].
Platelet Activation via ILRs
The most widely studied ILR in terms of signaling is GPVI, while less attention has been paid to the podoplanin receptor C-type lectin-like receptor 2 (CLEC2) and the IgG receptor FcγRIIa. In platelets, signaling via these ILRs relies on a tyrosine phosphorylation cascade starting at the (hemi-) ITAM motif YxxI/L [Citation39]. Tyrosine phosphorylation of this domain creates a docking site for SH2-containing proteins of the Src kinase family, which mediate Syk phosphorylation-ensuing activation of PLCγ and PI3K isoforms (). Positive feedback loops mediated by a crosstalk between the small-GTPases Rap1b and Rac1 appear to be critical for a full ITAM-induced platelet activation [Citation31].
As further detailed below, these ILRs, in particular, the collagen/fibrin receptor GPVI, have a well-established enhancing effect on the GPCR-induced responses by thrombin, ADP, and thromboxane A2 [Citation1]. Exemplary is the platelet procoagulant response mediated by PS exposure, requiring the combined collagen and thrombin stimulation [Citation5,Citation40,Citation41].
Glycoprotein VI (GPVI)
GPVI is uniquely expressed by megakaryocytes and platelets and requires co-expression with the ITAM-bearing co-receptor Fc receptor γ-chain (FcRγ) [Citation42]. It is the central signaling receptor of collagen, collagen-related peptides (CRP), and the snake venom convulxin [Citation43,Citation44]. The strength of the GPVI signal in response to collagen ligands is regulated by receptor clustering [Citation45]. Recent overviews of mouse studies indicate that GPVI is one of the few platelet-signaling proteins that contribute to murine arterial thrombosis with only a minor role in hemostasis [Citation46]. This feature makes GPVI an attractive target for the development of improved anti-platelet drugs to prevent thrombosis. Indeed, GPVI-deficient patients described so far present with no more than a minor bleeding tendency [Citation43,Citation47]. Interestingly, a genetic variant of GP6 was found to associate with an increased risk of venous thrombosis [Citation48].
Other (hem)-ITAM-Linked Receptors
The podoplanin receptor CLEC2 is highly expressed as a homodimer in platelets and megakaryocytes, and it is present at low levels in inflammatory dendritic cells and myeloid cells [Citation39,Citation49]. Mouse thrombosis studies suggest a blood-borne ligand, but this has not been identified yet [Citation50]. In comparison to the GPVI-linked FcRγ chain, the CLEC2 receptor contains a hemi-ITAM domain with a single YxxL sequence [Citation51], but its signaling pathway shows considerable overlap with that of GPVI. Besides a role in arterial thrombosis, CLEC2 is also involved in the maintenance of lymphatic vascular integrity and inflammation [Citation39,Citation49]. When stimulated by the snake venom rhodocytin, CLEC2 appeared to have a no more than limited role in platelet procoagulant activity [Citation52]. This is in agreement with the reported moderate Ca2+ rise induced by CLEC2 activation [Citation53].
The FcγRIIa receptor (also named CD32a) acts as a low-affinity receptor for the constant fragment of immunoglobulin G [Citation54]. Upon dimerization it allows platelets to bind to IgG immune complexes, resulting in platelet activation via a similar tyrosine kinase cascade as GPVI and ensuing Ca2+ mobilization [Citation55]. A role of the platelet FcγRIIa is known for a range of pathologies, such as immune thrombocytopenia, heparin-induced thrombocytopenia, bacterial infections, and cancer [Citation54].
Synergistic Platelet Calcium Signaling via GPCRs and ILRs
Platelets respond in a context-dependent manner to the different stimuli to which they are exposed [Citation2,Citation56]. Accordingly, when these cells are stimulated through GPCR (e.g., with thrombin, autocrine ADP, thromboxane A2) and through ILR (e.g., with collagen) this results in additive responses because of the complementarity of both types of signaling pathways [Citation1]. A key quantifiable and integratory signal of GPCR plus ILR stimulation is the rise in cytosolic-free Ca2+, which then feeds forward to other responses such as pseudopod formation, integrin αIIbβ3 activation, granular secretion, procoagulant activity, and formation of contracting aggregates [Citation1].
A notable mechanism of Ca2+ signal integration is provided by the different PLC isoforms that become activated through GPCRs (PLCβ2/3) and ILRs (PLCγ2) [Citation57,Citation58]. Both PLC types hydrolyze phosphatidylinositol 4,5-biphosphate into diacylglycerol (DAG) and inositol 1,4,5-triphosphate (IP3) [Citation18,Citation59]. The messenger IP3 binds to its receptors in the endoplasmic reticulum and promotes the release of stored Ca2+ (). Feedback is provided by concomitant signals, including protein kinase C isoforms and CalDAG-GEFI [Citation31]. In a way not very well understood, the signaling route via PLCβ2/3 or PLCγ2 lead to a different shape of the IP3-induced Ca2+ mobilization and the related entry of extracellular Ca2+, which hence differ between GPCR and ILR agonists.
In single platelets, a marked difference between GPCR and ILR stimulation is the IP3-induced oscillatory, spiking Ca2+ rises in response to thrombin or ADP [Citation13], and the continued Ca2+ rises in response to collagen [Citation60] ( and B). The reason for this difference is still not well understood, but it likely involves a higher activity of Ca2+-ATPases (SERCAs), pumping back the released Ca2+ into the stores after each spike [Citation61], in the case of GPCR-stimulated platelets.
The agonist-induced regulation of Ca2+ entry channels in platelets is reviewed in detail elsewhere [Citation62]. In brief, following Ca2+ store depletion, the endoplasmic reticulum Ca2+ sensor STIM1 interacts with the store-operated Ca2+ entry (SOCE) channel Orai1, causing influx of Ca2+ from the extracellular medium (). Studies with genetically modified mice have shown that this SOCE pathway is more predominant upon ILR stimulation (collagen) than upon GPCR stimulation (thrombin) [Citation62]. Yet, patients with mutations in STIM1 or ORAI1 were found to suffer from immune deficiency and bleeding symptoms, linked to defects in GPCR signals and thrombus formation [Citation63]. An additional non-SOCE Ca2+ entry mechanism via the nonselective TRPC cation channels is stimulated by thrombin, with a significant role in PS exposure [Citation64]. In particular, co-stimulation with collagen and thrombin activates these TRPC, allowing entry of Na+ and Ca2+ [Citation64].
Figure 3. Schematic overview of the Ca2+ entry pathways and PS exposure. Left part GPCRs: Ca2+ depletion from the intracellular stores by GPCRs is mainly triggered through PLCβ-induced generation of IP3. The DAG mediates transient receptor potential channel 6 (TRPC6) opening, allowing extracellular Ca2+ entry. The rise in cytosolic Ca2+ results in activation of integrin αIIbβ3 via CalDAG-GEFI and Rap1b. Right part ILRs: ILR-induced Ca2+ depletion from the intracellular stores most strongly promotes STIM1-Orai1 channel coupling and activation by pumping Ca2+ ions inside the cytosol (store-operated calcium entry, SOCE). Mitochondrial depolarization and generation of mitochondrial permeability transition pore (MPTP) contributes to the increase of cytoplasmic Ca2+ levels. This high and sustained Ca2+ levels mediate anoctamin-6 (Ano6) scramblase activation and PS exposure, which also promotes ion channel opening and Na+, Cl− and water entry. Among other processes, calpains are activated resulting in the cleavage of several proteins and receptors. In both pathways, high intracellular Ca2+ levels activate sarco/endoplasmic reticulum Ca2+ ATPases (SERCA), which pumps Ca2+ back into the intracellular stores. Several channels in the plasma membrane such as plasma membrane Ca2+ ATPases (PMCA) and Na+/Ca2+ exchanger (NCX) also reducing the cytoplasmic Ca2+ levels (not shown). ROCK: Rho-associated protein kinase. For other abbreviations, see . This figure was created using BioRender.com
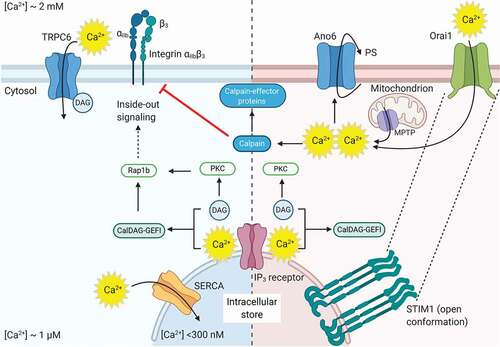
Furthermore, mitochondrial-stored Ca2+ can play a role upon dual platelet stimulation. Current understanding is that signals including elevated Ca2+ trigger the mitochondrial Ca2+ release via mitochondrial permeability transition pores, which results in the ‘supramaximal Ca2+ signals’ that are required for platelet PS exposure and procoagulant activity [Citation65,Citation66].
Another platelet ion channel highly selective for Ca2+ is provided by the P2X1 receptor for ATP, which also plays a regulatory role in thrombus formation [Citation67,Citation68]. P2X1 contributes to Ca2+ signaling and platelet function upon low-level stimulation by GPCRs and ILRs [Citation69]. The contribution of the P2X1 signal to PS exposure is less clear. Regarding secondary lowering of the Ca2+ signal, next to SERCA isoforms, also plasma membrane Ca2+-ATPases and Na+/Ca2+ exchangers play a role in this [Citation62], although it is unclear if these roles differ between platelet agonists.
Overall, it appears that the magnitude and duration of the Ca2+ signal in platelets is a function of their exposure to agonists, in which GPCR and ILR agonists together evoke the platelet procoagulant response. A differential exposure to agonists can also explain, on top of differences between platelets, why the platelets in a thrombus are heterogeneous in activation stages [Citation70,Citation71]. Herein, the population of most strongly activated platelets is the one showing PS exposure [Citation41,Citation56]. Taken together, the physiological consequences of procoagulant platelet formation can be summarized as: (i) limiting the thrombus growth by reducing the adhesive function of platelets, (ii) enhancing the coagulation by thrombin and fibrin formation, and (iii) a thrombin-induced stabilization and contraction of the platelet plug.
Anoctamin-6 and the Platelet Procoagulant Response
The channel anoctamin-6 is a multi-spanning integral membrane protein, coded by the gene ANO6 (TMEM16F). It is highly expressed in platelets and serves as a Ca2+-dependent ion channel as well as a channel for negatively charged phospholipids like PS [Citation6,Citation72]. Whereas first papers considered anoctamin-6 as a channel permeable to both monovalent and divalent cations, including Cl−, Na+ and Ca2+ [Citation73], later reports emphasized the high Ca2+-dependent Cl− conductance [Citation6,Citation74]. Interestingly, recent structural and inter-species analyses suggest that the ion permeation occurs independently of phospholipid scrambling, i.e. PS exposure [Citation74,Citation75]. However, the ion channel activity is required for the anoctamin-6-dependent ballooning of platelets that accompanies PS exposure [Citation76].
Dysfunctional mutations in ANO6 are causative for the Scott syndrome (), resulting in a mild bleeding phenotype in mouse and man [Citation82–84]. To date, only six Scott patients have been reported, who only bleed after major trauma (surgery) without easy bruising from superficial cuts [Citation77].
Table I. Genetic pathologies altering platelet PS exposure. Abbreviations: ΔCRAC, combined immunodeficiency due to CRAC channel dysfunction; EV, extracellular vesicle; WAS, Wiskott–Aldrich syndrome
Studies with human and mouse platelets have confirmed the role of anoctamin-6 in the procoagulant responses upon sustained intracellular Ca2+ rises, such as induced by the co-stimulation of collagen and thrombin () or Ca2+ ionophores. Such platelets swell due to anoctamin-6-mediated influx of Cl− and (in)directly Na+, forming balloon-like structures, and simultaneously expose procoagulant PS due to phospholipid scramblase activity. Both types of responses are annulled in platelets from Scott syndrome patients and whole-body or platelet-specific Ano6−/− mice [Citation76,Citation83,Citation84].
In platelets from at least two Scott patients, a reduced collagen/thrombin-induced activity of the Ca2+-dependent protease calpain was found, which can explain lower microvesicle formation and lower intracellular cleavage of integrin αIIbβ3 and talin in these platelets [Citation14,Citation83,Citation85]. For the ScottUK patient, this could be linked to a decreased expression of one of the calpain isoforms [Citation86]. Of relevance, in platelet-specific Ano6−/−- mice, the hemostatic and thrombotic reactions were reduced, thus confirming that platelet PS exposure is a regulatory factor in blood clotting in vivo [Citation84].
As indicated before, a condition par excellence that induces anoctamin-6 mediated PS exposure and procoagulant activity is the activation of platelets by collagen plus thrombin (next to Ca2+ ionophore), resulting in ‘supramaximal’ high Ca2+ rises [Citation5,Citation87]. These high-Ca2+ PS-exposing platelets are furthermore characterized by a calpain-mediated cleavage and inactivation of integrin αIIbβ3 [Citation88], the extracellular cleavage of GPVI and GPIbα [Citation89], and a greatly enhanced binding ability of Gla-domain containing coagulation factors [Citation18,Citation90]. This assembly of coagulation factors causes a magnitude increase in the formation of factor Xa (tenase complex) and thrombin (prothrombinase complex) at the platelet surface, thereby strongly enhancing thrombin generation and the clotting process [Citation1,Citation52,Citation91]. Since the ballooned PS-exposing platelets can collect a surrounding protein (fibrin) coat, they were also designated as COAT platelets (COllagen And Thrombin) [Citation92,Citation93]. Given the major morphological changes of the ballooned platelets, it is not a surprise that anoctamin-6 has a strong regulatory effect on the platelet neo-N-terminal and phospho-proteomes upon collagen/thrombin activation [Citation86]. With the risk of popularizing, for these non-dead platelets, the term ‘zombie’ might be more appropriate than ‘necrotic.’
On the other hand, PS exposure of platelets can also be induced by a different pathway, acting independently of anoctamin-6 and elevated Ca2+ [Citation94,Citation95]. The latter pathway is triggered by the apoptotic stimulus ABT-737, relies on caspase activation and is accompanied by a different set of intracellular proteolytic events [Citation86]. The responsible PS-exposing enzyme most likely is the phospholipid ‘scramblase’ XKR8 [Citation96], the Ca2+ dependence of which is unclear. As summarized elsewhere, mouse studies have shown that many other signaling proteins can modulate collagen/thrombin-induced PS exposure [Citation97]. However, in the vast majority of these are proteins that primarily alter the Ca2+ signal. As shown in , the clinical relevance of the altered PS exposure in patients with Stormorken, Wiskott-Aldrich or Calcium Release-Activated Channel (CRAC) syndromes lies in bleeding symptoms and thrombocytopenia as a result of alterations in Ca2+ signaling proteins.
High-Throughput Screening to Find New Inhibitors Affecting Platelet Calcium Signaling and Procoagulant Activity
High-throughput screening (HTS) of small compounds or biomolecules is an established technique for the discovery of lead molecules to produce commercialized agents after several rounds of drug development [Citation98]. So far, only a few high-throughput approaches have been used to search for platelet-related Ca2+ signal inhibitors (). A first example is a well-plate based high-throughput approach employing Drosophila cell lines to identify novel inhibitors of the Orai1 CRAC channels [Citation99]. Other examples are the use of HEK cells for identification of the novel PAR4 antagonist BMS-986120 [Citation23], and for the search for a small molecule inhibitor of the nonselective Ca2+ influx channel TRPC6 [Citation100]. In the platelet field, pharmaceutical screening approaches discriminating between GPCR- and ILR-induced Ca2+ signaling are still absent. In the reported studies of , platelets were used only after initial screening.
Table II. Used approaches of high-throughput screening for platelet-related Ca2+ research. Abbreviations: HEK, human embryonic kidney; PP1cα, protein phosphatase 1catalytic subunit; TG, thrombin generation
In this context, it is of interest to make use of the high and sustained Ca2+ rise that triggers PS exposure for a high-throughput screening setting of platelets to find novel molecules interfering with procoagulant activity. Finding inhibitors that block the appearance of procoagulant platelets may reduce the risk of thrombotic events, while preserving other populations of adhesive, secreting and aggregating platelets, thereby limiting the risk of bleeding. However, a fine-tuning inhibitory dosing would be needed to minimize the bleeding complications, such as seen in Scott syndrome patients. Besides this, it will also be interesting to find new procoagulant molecules to strengthen hemostasis in conditions, where this is needed.
Conclusion
Several pathological conditions in the spectrum of cardiovascular disease present with hyperreactive platelets and increased levels of procoagulant platelets [38, Baaten, Citation104Citation104Citation104Citation104Citation104Citation104Citation104 #53, 53]. However, useful inhibitors – with a clinical potential – targeting the high Ca2+ levels and consequent PS exposure in platelets stimulated by GPCR plus ILR agonists have not yet been reported. Therefore, studies on the role of procoagulant platelets in pathological conditions are needed to find novel targets that help in the development of inhibitors that prevent procoagulant platelet formation.
Disclosure of Interest
The authors declare no conflicts of interest.
Supplemental Material
Download MP4 Video (74.4 MB)Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Versteeg HH, Heemskerk JW, Levi M, Reitsma PS. New fundamentals in hemostasis. Physiol Rev 2013;93:327–358. doi:https://doi.org/10.1152/physrev.00016.2011
- Van der Meijden PE, Heemskerk JW. Platelet biology and functions: new concepts and future clinical perspectives. Nat Rev Cardiol 2019;16:166–179. doi:https://doi.org/10.1038/s41569-018-0110-0
- Kuijpers MJE, Munnix ICA, Cosemans JMEM, van Vlijmen BJ, Reutelingsperger CPM, Oude Egbrink MGA, Heemskerk JWM. Key role of platelet procoagulant activity in tissue factor- and collagen-dependent thrombus formation in arterioles and venules in vivo. Differential sensitivity to thrombin inhibition. Microcirculation 2008;15:269–282. doi:https://doi.org/10.1080/10739680701653517
- Kuijpers MJE, Gilio K, Reitsma S, Nergiz-Unal R, Prinzen L, Heeneman S, Lutgens E, van Zandvoort MAMJ, Nieswandt B, Oude Egbrink MGA, et al. Complementary roles of platelets and coagulation in thrombus formation on plaques acutely ruptured by targeted ultrasound treatment: a novel intravital model. J Thromb Haemost 2009;7:152–161. doi:https://doi.org/10.1111/j.1538-7836.2008.03186.x
- Heemskerk JW, Cosemans JM, van der Meijden PE. Platelets and coagulation. In: Gresele P, Kleiman NS, Lopez JA, Page CP, editors. Platelets in thrombotic and non-thrombotic disorders. Springer Nature, Switzerland. 2017. ISBN: 978-3-319-47460-1:447-462. doi:https://doi.org/10.1007/978-3-319-47462-5_32.
- Kunzelmann K, Nilius B, Owsianik G, Schreiber R, Ousingsawat J, Sirianant L, Wanitchakool P, Bevers EM, Heemskerk JWM. Molecular functions of anoctamin 6 (TMEM16F): a chloride channel, cation channel, or phospholipid scramblase? Pflügers Arch - Eur J Physiol 2014;466:407–414. doi:https://doi.org/10.1007/s00424-013-1305-1
- Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res 2006;99:1293–1304. doi:https://doi.org/10.1161/01.RES.0000251742.71301.16
- Jones ML, Norman JE, Morgan NV, Mundell SJ, Lordkipanidze M, Lowe GC, Daly ME, Simpson MA, Drake S, Watson SP, et al. Diversity and impact of rare variants in genes encoding the platelet G protein-coupled receptors. Thromb Haemost 2015;113:826–837. doi:https://doi.org/10.1160/TH14-08-0679
- Stockley J, Nisar SP, Leo VC, Sabi E, Cunningham MR, Eikenboom JC, Lethagen S, Schneppenheim R, Goodeve AC, Watson SP, et al. Identification and characterization of novel variations in platelet G-protein coupled receptor (GPCR) genes in patients historically diagnosed with type 1 von Willebrand disease. Plos One 2015;10:e0143913. doi:https://doi.org/10.1371/journal.pone.0143913
- Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV, Tam T, Coughlin SR. A dual thrombin receptor system for platelet activation. Nature 1998;6694:690–694. doi:https://doi.org/10.1038/29325
- Sambrano GR, Weiss EJ, Zheng YW, Huang W, Coughlin SR. Role of thrombin signalling in platelets in haemostasis and thrombosis. Nature 2001;413:74–78. doi:https://doi.org/10.1038/35092573
- Heemskerk JW, Feijge MA, Henneman L, Rosing J, Hemker HC. The Ca2+-mobilizing potency of a-thrombin and thrombin-receptor-activating peptide on human platelets. Concentration and time effects of thrombin-induced Ca2+ signaling. Eur J Biochem 1997;249:547–555. doi:https://doi.org/10.1111/j.1432-1033.1997.00547.x
- Heemskerk JW, Vis P, Feijge MA, Hoyland J, Mason WT, Sage SO. Roles of phospholipase C and Ca2+-ATPase in calcium responses of single, fibrinogen-bound platelets. J Biol Chem 1993;268:356–363.
- Munnix ICA, Harmsma M, Giddings JC, Collins PW, Feijge MAH, Comfurius P, Heemskerk JWM, Bevers EM. Store-mediated calcium entry in the regulation of phosphatidylserine exposure in blood cells from Scott patients. Thromb Haemost 2003;89:687–695. doi:https://doi.org/10.1055/S-0037-1613576
- Heemskerk JW, Vuist WM, Feijge MA, Reutelingsperger CP, Lindhout T. Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood 1997;90:2615–2625. doi:https://doi.org/10.1182/blood.V90.7.2615
- Covic L, Gresser AL, Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 2000;5458–5467. doi:https://doi.org/10.1021/bi9927078
- Shapiro MJ, Weiss EJ, Faruqi TR, Coughlin SR. Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J Biol Chem 2000;275:25216–25221. doi:https://doi.org/10.1074/jbc.M004589200
- Heemskerk JW, Mattheij N, Cosemans JM. Platelet-based coagulation: different populations, different functions. J Thromb Haemost 2013;11:2–11. doi:https://doi.org/10.1111/jth.12045
- French SL, Arthur JF, Lee H, Nesbitt WS, Andrews RK, Gardiner EE, Hamilton JR. Inhibition of protease-activated receptor 4 impairs platelet procoagulant activity during thrombus formation in human blood. J Thromb Haemost 2016;14:1642–1654. doi:https://doi.org/10.1111/jth.13293
- Jin J, Mao Y, Thomas D, Kim S, Daniel JL, Kunapuli SP. RhoA downstream of Gq and G12/13 pathways regulates protease-activated receptor-mediated dense granule release in platelets. Biochem Pharmacol 2009;77:835–844. doi:https://doi.org/10.1016/j.bcp.2008.11.017
- Norman JE, Cunningham MR, Jones ML, Walker ME, Westbury SK, Sessions RB, Mundell SJ, Mumford AD. Protease-activated receptor 4 variant p.Tyr157Cys reduces platelet functional responses and alters receptor trafficking. Arterioscler Thromb Vasc Biol 2016;36:952–960. doi:https://doi.org/10.1161/ATVBAHA.115.307102
- French SL, Thalmann C, Bray PF, Macdonald LE, Murphy AJ, Sleeman MW, Hamilton JR. A function-blocking PAR4 antibody is markedly antithrombotic in the face of a hyperreactive PAR4 variant. Blood Adv 2018;2:1283–1289. doi:https://doi.org/10.1182/bloodadvances.2017015552
- Wong PC, Seiffert D, Bird JE, Watson CA, Bostwick JS, Giancarli M, Allegretto N, Hua J, Harden D, Guay J, et al. Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci Transl Med 2017;9:371. doi:https://doi.org/10.1126/scitranslmed.aaf5294
- Gryka RJ, Buckley LF, Anderson SM. Vorapaxar: the current role and future directions of a novel protease-activated receptor antagonist for risk reduction in atherosclerotic disease. Drugs 2017;65–72. doi:https://doi.org/10.1007/s40268-016-0158-4
- Tourdot BE, Stoveken H, Trumbo D, Yeung J, Kanthi Y, Edelstein LC, Bray PF, Tall GG, Holinstat M. Genetic variant in human PAR 4 enhances thrombus formation resulting in resistance to antiplatelet therapeutics. Arterioscler Thromb Vasc Biol 2018;38:1632–1643. doi:https://doi.org/10.1161/ATVBAHA.118.311112
- Gachet C. Identification, characterization, and inhibition of the platelet ADP receptors. Int J Hematol 2001;74:375–381. doi:https://doi.org/10.1007/BF02982079
- Hechler B, Gachet C. P2 receptors and platelet function. Purinergic Signal 2011;3:293–303. doi:https://doi.org/10.1007/s11302-011-9247-6
- Leon C, Ravanat C, Freund M, Cazenave JP, Gachet C. Differential involvement of the P2Y1 and P2Y12 receptor in platelet procoagulant activity. Arterioscler Thromb Vasc Biol 2003;23:1941–1947. doi:https://doi.org/10.1007/s11302-011-9247-6
- Bye AP, Gibbins JM, Mahaut-Smith MP. Ca2+ waves coordinate purinergic receptor-evoked integrin activation and polarization. Sci Signal 2020;13:e7354. doi:https://doi.org/10.1126/scisignal.aav7354
- Hardy AR, Jones ML, Mundell SJ, Poole AW. Reciprocal cross-talk between P2Y1 and P2Y12 receptors at the level of calcium signaling in human platelets. Blood 2004;104:1745–1752. doi:https://doi.org/10.1182/blood-2004-02-0534
- Stefanini L, Roden RC, Bergmeier W. CalDAG-GEFI is at the nexus of calcium-dependent platelet activation. Blood 2009;114:2506–2514. doi:https://doi.org/10.1182/blood-2009-04-218768
- Mundell SJ, Rabbolini D, Gabrielli S, Chen Q, Aungraheeta R, Hutchinson JL, Kilo T, Mackay J, Ward CM, Stevenson W, et al. Receptor homodimerization plays a critical role in a novel dominant negative P2RY12 variant identified in a family with severe bleeding. J Thromb Haemost 2018;16:44–53. doi:https://doi.org/10.1111/jth.13900
- Janicki PK, Eyileten C, Ruiz-Velasco V, Sedeek KA, Pordzik J, Czlonkowska A, Kurkowska-Jastrzebska I, Sugino S, Imamura-Kawasawa Y, Mirowska-Guzel D, et al. Population-specific associations of deleterious rare variants in coding region of P2RY1-P2RY12 purinergic receptor genes in large-vessel ischemic stroke patients. Int J Mol Sci 2017;18:2678. doi:https://doi.org/10.3390/ijms18122678
- Khan SU, Singh M, Valavoor S, Khan MU, Lone AN, Khan MZ, Khan MS, Mani P, Kapadia SR, Michos ED, et al. Dual antiplatelet therapy after percutaneous coronary intervention and drug-eluting stents: a systematic review and network meta-analysis. Circulation 2020;142:1425–1436. doi:https://doi.org/10.1161/CIRCULATIONAHA.120.046308
- Offermans S, Laugwitz KL, Spicher K, Schultz G. G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc Natl Acad Sci USA 1994;91:504–508. doi:https://doi.org/10.1073/pnas.91.2.504
- Hirata T, Kakizuka A, Ushikubi F, Fuse I, Okuma M, Narumiya S. Arg60 to Leu mutation of the human thromboxane A2 receptor in a dominantly inherited bleeding disorder. J Clin Invest 1994;94:1662–1667. doi:https://doi.org/10.1172/JCI117510
- Mundell SJ, Mumford A. TBXA2R gene variants associated with bleeding. Platelets 2018;29:739–742. doi:https://doi.org/10.1080/09537104.2018.1499888
- Swieringa F, Kuijpers MJ, Heemskerk JW, van der Meijden PE. Targeting platelet receptor function in thrombus formation: the risk of bleeding. Blood Rev 2014;28:9–21. doi:https://doi.org/10.1016/j.blre.2013.12.001
- Rayes J, Watson SP, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest 2019;129:12–23. doi:https://doi.org/10.1172/JCI122955
- Lecut C, Feijge MAH, Cosemans JMEM, Jandrot-Perrus M, Heemskerk JWM. Fibrillar type I collagens enhance platelet-dependent thrombin generation via glycoprotein VI with direct support of a2b1 but not aIIbb3 integrin. Thromb Haemost 2005;94:107–114. doi:https://doi.org/10.1160/TH04-12-0783
- Agbani EO, Poole AW. Procoagulant platelets: generation, function, and therapeutic targeting in thrombosis. Blood 2017;130:2171–2179. doi:https://doi.org/10.1182/blood-2017-05-787259
- Stegner D, Haining E, Nieswandt B. Targeting glycoprotein VI and the immunoreceptor tyrosine-based activation motif signaling pathway. Arterioscler Thromb Vasc Biol 2014;34:1615–1620. doi:https://doi.org/10.1161/ATVBAHA.114.303408
- Jandrot-Perrus M, Busfield S, Lagrue AH, Xiong X, Debili N, Chickering T, Le Couedic JP, Goodearl A, Dussault B, Fraser C, et al. Cloning, characterization, and functional studies of human and mouse glycoprotein VI: a platelet-specific collagen receptor from the immunoglobulin superfamily. Blood 2000;96:1798–1807. doi:https://doi.org/10.1182/blood.V96.5.1798
- Smethurst PA, Joutsi-Korhonen L, O’Connor MN, Wilson E, Jennings NS, Garner SF, Zhang Y, Knight CG, Dafforn TR, Buckle A, et al. Identification of the primary collagen-binding surface on human glycoprotein VI by site-directed mutagenesis and by a blocking phage antibody. Blood 2004;103:903–911. doi:https://doi.org/10.1182/blood-2003-01-0308
- Poulter NS, Pollitt AY, Owen DM, Gardiner EE, Andrews RK, Shimizu H, Ishikawa D, Bihan D, Farndale RW, Moroi M, et al. Clustering of glycoprotein VI (GPVI) dimers upon adhesion to collagen as a mechanism to regulate GPVI signaling in platelets. J Thromb Haemost 2017;15:549–564. doi:https://doi.org/10.1111/jth.13613
- Baaten CC, Meacham S, de Witt SM, Feijge MA, Adams DJ, Akkerman JW, Cosemans JM, Grassi L, Jupe S, Kostadima M, et al. A synthesis approach of mouse studies to identify genes and proteins in arterial thrombosis and bleeding. Blood 2018;132:e35–e46. doi:https://doi.org/10.1182/blood-2018-02-831982
- Nagy M, Perrella G, Dalby A, Becerra M, Garcia Quintanilla L, Pike J, Morgan N, Gardiner E, Heemskerk JW, Azocar L, et al. Flow studies on human GPVI-deficient blood under coagulating and non-coagulating conditions. Blood Adv 2020;4:2953–2961. doi:https://doi.org/10.1182/bloodadvances.2020001761
- Morange PE, Suchon P, Trégouët DA. Genetics of venous thrombosis: update in 2015. Thromb Haemost 2015;114:910–919. doi:https://doi.org/10.1160/TH15-05-0410
- Suzuki-Inoue K, Inoue O, Ding G, Nishimura S, Hokamura K, Eto K, Kashiwagi H, Tomiyama Y, Yatomi Y, Umemura K, et al. Essential in vivo roles of the C-type lectin receptor CLEC-2. Embryonic/neonatal lethality of CLEC-2-deficient mice by blood/lymphatic misconnections and impaired thrombus formation of CLEC-2-deficient platelets. J Biol Chem 2010;285:24494–24507. doi:https://doi.org/10.1074/jbc.M110.130575
- May F, Hagedorn I, Pleines I, Bender M, Vögtle T, Eble J, Elvers M, Nieswandt B. CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood 2009;114:3464–3472. doi:https://doi.org/10.1182/blood-2009-05-222273
- Hughes CE, Navarro-Nunez L, Finney BA, Mourao-Sa D, Pollitt AY, Watson SP. CLEC-2 is not required for platelet aggregation at arteriolar shear. J Thromb Haemost 2010;8:2328–2332. doi:https://doi.org/10.1111/j.1538-7836.2010.04006.x
- Brouns SLN, van Geffen JP, Campello E, Swieringa F, Spiezia L, van Oerle R, Provenzale I, Verdoold R, Farndale RW, Clemetson KJ, et al. Platelet-primed interactions of coagulation and anticoagulation pathways in flow-dependent thrombus formation. Sci Rep 2020;10:in press. doi:https://doi.org/10.1038/s41598-020-68438-9
- Martyanov AA, Balabin FA, Dunster JL, Panteleev MA, Gibbins JM, Sveshnikova AN. Control of platelet CLEC-2-mediated activation by receptor clustering and tyrosine kinase signaling. Biophys J 2020;118:2641–2655. doi:https://doi.org/10.1016/j.bpj.2020.04.023
- Qiao J, Al-Tamimi M, Baker RI, Andrews RK, Gardiner EE. The platelet Fc receptor, FcgammaRIIa. Immunol Rev 2015;268:241–252. doi:https://doi.org/10.1111/imr.12370
- Lee RH, Bergmeier W. Platelet immunoreceptor tyrosine-based activation motif (ITAM) and hemITAM signaling and vascular integrity in inflammation and development. J Thromb Haemost 2016;14:645–654. doi:https://doi.org/10.1111/jth.13250
- Baaten CC, Ten Cate H, van der Meijden PE, Heemskerk JW. Platelet populations and priming in hematological diseases. Blood Rev 2017;31:389–399. doi:https://doi.org/10.1016/j.blre.2017.07.004
- Lian L, Wang Y, Draznin J, Eslin D, Bennett JS, Poncz M, Wu D, Abrams CS. The relative role of PLCb and PI3Kg in platelet activation. Blood 2005;106:110–117. doi:https://doi.org/10.1182/blood-2004-05-2005
- Suzuki-Inoue K, Inoue O, Frampton J, Watson SP. Murine GPVI stimulates weak integrin activation in PLCg2 -/- platelets: involvement of PLCg1 and PI-kinase. Blood 2003;102:1367–1373. doi:https://doi.org/10.1182/blood-2003-01-0029
- Varga-Szabo D, Braun A, Nieswandt B. STIM1 and Orai1 in platelet function. Cell Calcium 2011;50:70–278. doi:https://doi.org/10.1111/j.1538-7836.2009.03455.x
- Heemskerk JW, Siljander P, Vuist WM, Breikers G, Reutelingsperger CP, Barnes MJ, Knight CG, Lassila R, Farndale RW. Function of glycoprotein VI and integrin a2b1 in the procoagulant response of single, collagen-adherent platelets. Thromb Haemost 1999;81:782–792. doi:https://doi.org/10.1055/S-0037-1614571
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 2003;4:517–529. doi:https://doi.org/10.1038/nrm1155
- Mammadova-Bach E, Nagy M, Heemskerk JW, Nieswandt N, Braun A. Store-operated calcium entry in blood cells in thrombo-inflammation. Cell Calcium 2019;77:39–48. doi:https://doi.org/10.1016/j.ceca.2018.11.005
- Nagy M, Mastenbroek TG, Mattheij NJ, De Witt S, Clemetson KJ, Kirschner J, Schulz A, Braun A, Cosemans JM, Zieger B, et al. Variable impairment of platelet functions in patients with severe, genetically linked immune deficiencies. Haematologica 2018;103:540–549. doi:https://doi.org/10.3324/haematol.2017.176974
- Harper MT, Camacho-Londono JE, Quick K, Camacho-Londono J, Flockerzi V, Phillipp SE, Birnbaumer L, Freichei M, Poole AW. Transient receptor potential channels function as a coincidence signal mediating phosphatidylserine exposure. Sci Signal 2013;6:ra50. doi:https://doi.org/10.1126/scisignal.2003701
- Jobe SM, Wilson KM, Leo L, Raimondi A, Molkentin JD, Lentz SR, DiPaola J. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008;111:1257–1265. doi:https://doi.org/10.1182/blood-2007-05-092684
- Abbasian N, Millington-Burgess SL, Chabra S, Malcor JD, Harper MT. Supramaximal calcium signaling triggers procoagulant platelet formation. Blood Adv 2020;14:154–164. doi:https://doi.org/10.1182/bloodadvances.2019000182
- Hechler B, Lenain N, Marchese P, Vial C, Heim V, Freund M, Cazenave JP, Cattaneo M, Ruggeri ZM, Evans R, et al. A role of the fast ATP-gated P2X1 cation channel in thrombosis of small arteries in vivo. J Exp Med 2003;198:661–667. doi:https://doi.org/10.1182/bloodadvances.2019000182
- Oury C, Kuijpers MJ, Toth-Zsamboki E, Bonnefoy A, Danloy S, Vreys I, Feijge MA, De Vos R, Vermylen J, Heemskerk JW, et al. Overexpression of the platelet P2X1 ion channel in transgenic mice generates a novel prothrombotic phenotype. Blood 2003;101:3969–3976. doi:https://doi.org/10.1182/blood-2002-10-3215
- Fung CY, Cendana C, Farndale RW, Mahaut-Smith MP. Primary and secondary agonists can use P2X1 receptors as a major pathway to increase intracellular Ca2+ in the human platelet. J Thromb Haemost 2007;5:910–917. doi:https://doi.org/10.1111/j.1538-7836.2007.02525.x
- Munnix IC, Kuijpers MJ, Auger JM, Thomassen CM, Panizzi P, van Zandvoort MA, Rosing J, Bock PE, Watson SP, Heemskerk JW. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation. Regulation by transient integrin activation. Arterioscler Thromb Vasc Biol 2007;27:2484–2490. doi:https://doi.org/10.1161/ATVBAHA.107.151100
- Stalker TJ, Traxler EA, Wu J, Wannemacher KM, Cermignano SL, Voronov R, Diamond SL, Brass LF. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood 2013;121:1875–1885. doi:https://doi.org/10.1182/blood-2012-09-457739
- Fujii T, Sakata A, Nishimura S, Eto K, Nagata S. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc Natl Acad Sci USA 2015;112:12800–12805. doi:https://doi.org/10.1073/pnas.1516594112
- Yang H, Kim A, David T, Palmer D, Jin T, Tien J, Huang F, Cheng T, Coughlin SR, Jan YN, et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 2012;151:111–122. doi:https://doi.org/10.1016/j.cell.2012.07.036
- Taylor KA, Mahaut-Smith MP. A major interspecies difference in the ionic selectivity of megakaryocyte Ca2+-activated channels sensitive to the TMEM16F inhibitor CaCCinh-A01. Platelets 2019;30:962–966. doi:https://doi.org/10.1080/09537104.2019.1595560
- Ye W, Han TW, He M, Jan YN, Jan LY. Dynamic change of electrostatic field in TMEM16F permeation pathway shifts its ion selectivity. Elife 2019;8:e45187. doi:https://doi.org/10.7554/eLife.45187.001
- Agbani EO, van den Bosch MT, Brown E, Williams CM, Mattheij NJ, Cosemans JM, Collins PW, Heemskerk JW, Hers I, Poole AW. Coordinated membrane ballooning and procoagulant spreading in human platelets. Circulation 2015;132:1414–1424. doi:https://doi.org/10.1161/CIRCULATIONAHA.114.015036
- Millington-Burgess SL, Harper MT. Gene of the issue: ANO6 and Scott syndrome. Platelets 2020;31:964–967. doi:https://doi.org/10.1080/09537104.2019.1693039
- Nurden AT, Nurden P. Inherited disorders of platelet function: selected updates. J Thromb Haemost 2015;13:S2–S9. doi:https://doi.org/10.1111/jth.12898
- Misceo D, Holmgren A, Louch WE, Holme PA, Mizobuchi M, Morales RJ, De Paula AM, Stray-Pedersen A, Lyle R, Dalhus B, et al. A dominant STIM1 mutation causes Stormorken syndrome. Hum Mutat 2014;35:556–564. doi:https://doi.org/10.1002/humu.22544
- Lemahieu V, Gastier JM, Francke U. Novel mutations in the Wiskott-Aldrich syndrome protein gene and their effects on transcriptional, translational, and clinical phenotypes. Hum Mutat 1999;14:54–66. doi:https://doi.org/10.1002/(SICI)1098-1004(1999)14:1<54::AID-HUMU7>3.0.CO;2-E
- Feske S. CRAC channels and disease: from human CRAC channelopathies and animal models to novel drugs. Cell Calcium 2019;80:112–116. doi:https://doi.org/10.1016/J.CECA.2019.03.004
- Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 2010;468:834–838. doi:https://doi.org/10.1038/nature09583
- Mattheij NJA, Braun A, van Kruchten R, Castoldi E, Pircher J, Baaten CCFMJ, Wülling M, Kuijpers MJE, Köhler R, Poole AW, et al. Survival protein anoctamin-6 controls multiple platelet responses including phospholipid scrambling, swelling and protein cleavage. Faseb J 2016;30:727–737. doi:https://doi.org/10.1096/fj.15-280446
- Baig AA, Haining EJ, Geuss E, Beck S, Swieringa F, Wanitchakool P, Schuhmann MK, Stegner D, Kunzelmann K, Kleinschnitz C, et al. TMEM16F-mediated platelet membrane phospholipid scrambling is critical for hemostasis and thrombosis but not thromboinflammation in mice. Arterioscler Thromb Vasc Biol 2016;36:2152–2157. doi:https://doi.org/10.1161/ATVBAHA.116.307727
- Toti F, Satta N, Fressinaud E, Meyer D, Freyssinet JM. Scott syndrome, characterized by impaired transmembrane migration of procoagulant phosphatidylserine and hemorrhagic complications, is an inherited disorder. Blood 1996;87:1409–1415. doi:https://doi.org/10.1182/blood.V87.4.1409.bloodjournal8741409
- Solari FA, Mattheij NJ, Burkhart JM, Swieringa F, Collins PW, Cosemans JM, Sickmann A, Heemskerk JW, Zahedi RP. Combined quantification of the global proteome, phosphoproteome and protein cleavage to characterize altered platelet functions in the human Scott syndrome. Mol Cell Proteomics 2016;15:3154–3169. doi:https://doi.org/10.1074/mcp.M116.060368
- Bevers EM, Comfurius P, van Rijn JL, Hemker HC, Zwaal RF. Generation of prothrombin-converting activity and the exposure of phosphatidylserine at the outer surface of platelets. Eur J Biochem 1982;122:429–436. doi:https://doi.org/10.1111/j.1432-1033.1982.tb05898.x
- Mattheij NJ, Gilio K, van Kruchten R, Jobe SM, Wieschhaus AJ, Chishti AH, Collins P, Heemskerk JW, Cosemans JM. Dual mechanism of integrin αIIbβ3 closure in procoagulant platelets. J Biol Chem 2013;288:13325–13336. doi:https://doi.org/10.1074/jbc.M112.428359
- Baaten CC, Swieringa F, Misztal T, Mastenbroek TG, Feijge MA, Bock PE, Donners MM, Collins PW, Li R, van der Meijden PE, et al. Platelet heterogeneity in activation-induced glycoprotein shedding: functional effects. Blood Adv 2018;2:2320–2331. doi:https://doi.org/10.1182/bloodadvances.2017011544
- Panteleev MA, Ananyeva NM, Greco NJ, Ataullakhanov FI. Two subpopulations of thrombin-activated platelets differ in their binding of the components of the intrinsic factor Xa-activating complex. J Thromb Haemost 2005;3:2545–2553. doi:https://doi.org/10.1111/J.1538-7836.2005.01616.X
- Reddy EC, Rand ML. Procoagulant phosphatidylserine-exposing platelets in vitro and in vivo. Front Cardiovasc Med 2020;7:15. doi:https://doi.org/10.3389/fcvm.2020.00015
- Dale GL, Friese P, Batar P, Hamilton SF, Reed GL, Jackson KW, Clemetson KJ, Alberio L. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature 2002;415:175–179. doi:https://doi.org/10.1038/415175a
- Alberio L, Ravanat C, Hechler B, Mangin PH, Lanza F, Gachet C. Delayed-onset of procoagulant signalling revealed by kinetic analysis of COAT platelet formation. Thromb Haemost 2017;117. doi:https://doi.org/10.1160/TH16-09-0711
- Schoenwaelder SM, Yuan Y, Josefsson EC, White MJ, Tao Y, Mason KD, O’Reilly LA, Henley KJ, Ono A, Hsiao S, et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood 2009;114:663–666. doi:https://doi.org/10.1182/blood-2009-01-200345
- Van Kruchten R, Mattheij NJA, Saunders C, Feijge M, Swieringa F, Wolfs JLM, Collins PW, Heemskerk JWM, Bevers EM. Both TMEM16F-dependent and TMEM16F-independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood 2013;121:1850–1857. doi:https://doi.org/10.1182/blood-2012-09-454314
- Sakuragi T, Kosako H, Nagata S. Phosphorylation-mediated activation of mouse Xkr8 scramblase for phosphatidylserine exposure. Proc Natl Acad Sci USA 2019;116:2907–2912. doi:https://doi.org/10.1073/pnas.1820499116
- De Witt S, Verdoold R, Cosemans JM, Heemskerk JW. Insights into platelet-based control of coagulation. Thromb Res 2014;133(Suppl 2):S139–148. doi:https://doi.org/10.1016/S0049-3848(14)50024-2
- Mayr LM, Bojanic D. Novel trends in high-throughput screening. Curr Opin Pharmacol 2009;9:580–588. doi:https://doi.org/10.1016/j.coph.2009.08.004
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006;11:179–185. doi:https://doi.org/10.1038/nature04702
- Häfner S, Urban N, Schaefer M. Discovery and characterization of a positive allosteric modulator of transient receptor potential canonical 6 (TRPC6) channels. Cell Calcium 2019;78:26–34. doi:https://doi.org/10.1016/j.ceca.2018.12.009
- Pradhan S, Khatlani T, Nairn AC, Vijayan KV. The heterotrimeric G protein Gβ 1 interacts with the catalytic subunit of protein phosphatase 1 and modulates G protein-coupled receptor signaling in platelets. J Biol Chem 2017;292:13133–13142. doi:https://doi.org/10.1074/jbc.M117.796656
- Dowal L, Sim DS, Dilks JR, Blair P, Beaudry S, Denker BM, Koukos G, Kuliopulos A, Flaumenhaft R. Identification of an antithrombotic allosteric modulator that acts through helix 8 of PAR1. Proc Natl Acad Sci USA 2011;108:2951–2956. doi:https://doi.org/10.1073/pnas.1014863108
- Gerotziafas GT, Elalamy I, Depasse F, Perzborn E, Samama MM. In vitro inhibition of thrombin generation, after tissue factor pathway activation, by the oral, direct factor Xa inhibitor rivaroxaban. J Thromb Haemost 2007;5:886–888. doi:https://doi.org/10.1111/J.1538-7836.2007.02429.X
- Baaten CC, Ten Cate H, van Der Meijden PE, Heemskerk JW. Platelet populations and priming in hematological diseases. Blood Rev 2017;31:389-399. doi: https://doi.org/10.1016/j.blre.2017.07.004