
Abstract
Kv1.3 is a voltage-gated K+-selective channel with roles in immunity, insulin-sensitivity, neuronal excitability and olfaction. Despite being one of the largest ionic conductances of the platelet surface membrane, its contribution to platelet function is poorly understood. Here we show that Kv1.3-deficient platelets display enhanced ADP-evoked platelet aggregation and secretion, and an increased surface expression of platelet integrin αIIb. In contrast, platelet adhesion and thrombus formation in vitro under arterial shear conditions on surfaces coated with collagen were reduced for samples from Kv1.3−/- compared to wild type mice. Use of collagen-mimetic peptides revealed a specific defect in the engagement with α2β1. Kv1.3−/- platelets developed significantly fewer, and shorter, filopodia than wild type platelets during adhesion to collagen fibrils. Kv1.3−/- mice displayed no significant difference in thrombus formation within cremaster muscle arterioles using a laser-induced injury model, thus other pro-thrombotic pathways compensate in vivo for the adhesion defect observed in vitro. This may include the increased platelet counts of Kv1.3−/- mice, due in part to a prolonged lifespan. The ability of Kv1.3 to modulate integrin-dependent platelet adhesion has important implications for understanding its contribution to normal physiological platelet function in addition to its reported roles in auto-immune diseases and thromboinflammatory models of stroke.
Introduction
Kv1.3 is a ubiquitously-expressed voltage-gated K+ channel with recognized roles in several physiological responses, including T cell activation, olfaction, and peripheral insulin sensitivity [Citation1–3]. Furthermore, Kv1.3 inhibition has been proposed as a treatment for auto-immune diseases, obesity, neuroinflammation and other conditions [Citation4–6]. In addition to its cell surface expression, this transmembrane protein is also located in the outer mitochondrial membrane where it has been linked to regulation of apoptosis and may therefore be a target for the treatment of cancer [Citation7,Citation8].
Voltage-gated potassium-selective channels displaying rapid activation and slow inactivation typical of Kv1.3 were first observed via patch clamp recordings in mammalian platelets by Maruyama[Citation9]. Experiments in human platelets and murine megakaryocytes later demonstrated that these channels, encoded by KCNA3, are responsible for setting the resting membrane potential of approximately −50 to −60 mV [Citation10–12]. Subsequent reports using megakaryocytes from other mammalian species further support these conclusions [Citation13–15]. Kv1.3−/- mice demonstrate that loss of the channel reduces platelet agonist-evoked Ca2+ responses and increases the circulating platelet count [Citation11,Citation16]. However, major questions remain regarding the overall impact of Kv1.3 on platelet responses and the underlying mechanisms. Using Kv1.3-deficient mice and a range of in vitro and in vivo assays, we have explored the contribution of this voltage-gated channel to platelet function and lifespan. A key finding is that Kv1.3 contributes to collagen-dependent adhesion and motility through interaction with the integrin α2β1. This advances our understanding of how Kv1.3 can contribute to function in platelets and other cell types, particularly within the immune system.
Methods
Reagents and antibodies
Antibodies for analysis of platelet surface antigens included FITC-conjugated rat anti-mouse GPIbα (CD42b, Xia.G5), GPIbβ (CD42c, Xia.C3), GPV (CD42d, Gon.C2) and rat anti-mouse isotype control (P190-1) from Emfret Analytics (Eibelstadt, Germany). Antibodies against integrin chains were FITC-conjugated α2 (CD49b, Ha 1/29), αIIb (CD41, MWReg30), β1 (CD29, Ha2/5), β3 (CD61, 2 C9.G2) and isotype controls from BD Biosciences (Wokingham, UK). Platelet α-granule secretion was measured using anti-P-selectin-FITC (CD62P, Wug.E9) and IgG isotype control, (Emfret Analytics). Horm collagen (type I fibrils from equine tendon) was obtained from Alere (Stockport, Cheshire, UK) and the collagen peptides CRP-XL: crosslinked GCO(GPO)10GCOG-amide, VWF-III: GPC(GPP5)GPRGQOGVMGFO(GPP)5GPC-amide, and GFOGER: GPC(GPP5)GFOGER(GPP5)GPC-amide, were from CambCol Laboratories (Ely, Cambs, UK). Fibrinogen, 3,3ʹ dihexyloxacarbocyanine iodide (DiOC6), prostaglandin E (PGE1), apyrase (type VII), ADP, and hirudin were all purchased from Sigma-Aldrich (Dorset, UK). FM®1-43 lipophilic styryl dye was from Molecular Probes (Life Technologies, Paisley, UK) and Phe-Pro-Arg-chloromethylketone (PPACK) from Hematologic Technologies Incorporated (Vermont, USA). DyLight® 649-conjugated anti-GPIbβ antibody (Emfret Analytics) was used for in vivo thrombus formation experiments.
Animals and murine blood sampling
The generation of Kv1.3-deficient mice has been described previously[Citation17]. These were backcrossed against C57BL/6 (Charles River, UK) and Kv1.3−/- mice confirmed by genotyping (). C57BL/6 mice matched for age and sex were purchased from Charles River, UK to represent wild-type (WT) controls. Experiments were carried out using mice of mixed gender. Blood was collected from the inferior vena cava of terminally isoflurane-anesthetised mice into 40 µM PPACK for whole blood in vitro studies of platelet adhesion under conditions of arterial flow, or acid citrate dextrose (ACD; 85 mM trisodium citrate, 78 mM citric acid and 111 mM glucose) for all assays using washed platelets (described below). All procedures were carried out in accordance with local and Home Office guidelines and approved Institutonal Animal Welfare Ethics Review Boards.
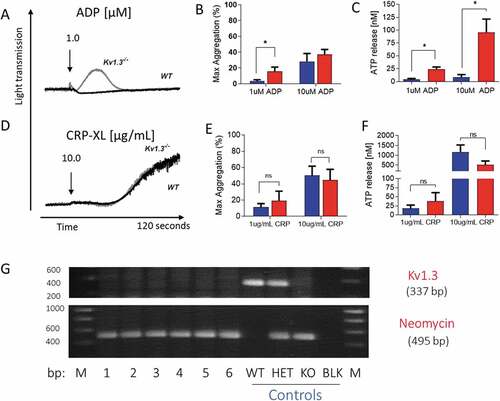
Figure 1. Platelet aggregation and secretion in wild type and Kv1.3−/- mice. (A,D) Representative traces of platelet aggregation in response to (A) 1 µM ADP, and (D) 10 µg/mL CRP-XL (black line, C57BL/6 WT and gray line, Kv1.3−/- (note that the WT and Kv1.3−/- aggregation traces completely overlap in D). (B, E) Mean percent peak aggregation of washed murine platelets in response to ADP (1 and 10 μM, B) and CRP-XL (1 and 10 μg/mL, E) is shown for WT (blue) and Kv1.3−/- (red) mice (mean ± SEM, n = 5). (C, F) Platelet dense granule secretion measured by analysis of ATP release in response to ADP (1 and 10 μM, C) and CRP-XL (1 and 10 μg/mL, F). Values are the mean ± SEM, n = 5; *P < .05, **P < .01, ns = not significant. (G) Representative gel showing the genotyping of C57BL/6 WT, heterozygous, and Kv1.3−/- mice. WT display 337-bp WT band only, Kv1.3−/- display the 495-bp neomycin band only, and heterozygous mice display both bands. The numbers across the bottom of the lanes denote individual samples and controls: bp = base pairs, M = molecular marker, lanes numbered 1–6 contain samples from Kv1.3−/- mice, followed by control samples from wild type (WT), heterozygous (HET) and Kv1.3−/- (KO) mice; BLK = PCR negative control, and M = molecular marker.
Preparation of washed platelets
Whole blood drawn into ACD was centrifuged at 300 g, 3 minutes, the platelet-rich plasma (PRP) removed and re-centrifuged at 200 g, 2 minutes to pellet remaining red blood cells. The platelet suspension was supplemented with PGE1 (100 ng/mL) and apyrase (0.32 U/mL), and centrifuged at 1000 g, 10 minutes. The platelet pellet was washed in normal platelet saline (NPS: 145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM D-glucose, 10 mM HEPES ~3.5 mM NaOH, pH 7.35) supplemented with 100 ng/mL PGE1 and apyrase (0.32 U/mL), and following centrifugation at 1000 g, was resuspended to original volume in NPS, and platelets adjusted to a concentration of 4 x 108/mL.
Flow cytometric analysis of platelet surface glycoproteins
Flow cytometry was used to measure the density of platelet surface glycoproteins. Washed platelets were diluted in NPS (1:10) and incubated for 15 minutes at room temperature with antibody or isotype control as per the manufacturer’s protocol. Platelet suspensions were diluted a further 1:10 in 0.2% formylsaline and analyzed by flow cytometer (BD Facscanto II; BD Biosciences, Wokingham, UK), gating the platelet population initially by size (Forward Scatter, FSC) and granularity (Side Scatter, SSC), followed by detection of mean fluorescent intensity of each surface antigen. All flow cytometry data was analyzed using Kaluza software (Beckman Coulter). Note that the values are reported as arbitrary fluorescence units since the signal depends upon the instrument gain and sensitivity.
Aggregation and secretion studies
Turbidimetric measurement of platelet aggregation was performed using a model 400 lumi-aggregometer (Chronolog, Manchester, UK). Washed platelet suspensions were stirred for 3 minutes at 37°C, then fibrinogen (100 µg/mL), calcium chloride (2 mM) and agonist added (ADP or CRP-XL), and platelet aggregation recorded for 2 minutes. In parallel, ATP secretion from dense granules was measured using the CHRONO-LUME® luciferin;luciferase assay according to the manufacturer’s guidelines.
Whole blood perfusion experiments
Whole blood was collected into 40 µM PPACK, and platelets loaded with 1 μM DiOC6for 20 minutes. Blood was perfused over coverslips coated with collagen (100 µg/mL), collagen peptide (100 µg/mL) or fibrinogen (200 µg/mL) at a shear rate of 1800 s−1 for collagen and collagen peptides, and 800 s−1 for fibrinogen. Thrombi were imaged by collection of a z-series of images acquired with an Olympus FV1000 confocal microscope at 3 separate fields per coverslip and analyzed in Image-J v1.49 (National Institutes of Health). Percent surface coverage, mean thrombus height and mean thrombus volume was calculated as described previously[Citation18]. For study of platelet morphology, images of immobilized platelets were recorded at 30 minutes, and analyzed using Image-J.
Platelet motility studies
Washed platelets were incubated with FM®1-43 (5 µM) and exposed to collagen-coated coverslips for 30 minutes in the absence of flow, and platelet movement and adhesion recorded in real-time on an Olympus FV1000 confocal microscope (excitation of FM®1-43 at 488 nm and emission at 550–650 nm). All experiments used a 60x oil immersion lens (UPLSAPO 60x, NA 1.35). The Image-J Manual Tracking plug-in was used to track the movement of platelets from each genotype as they attached and responded to the collagen fibers.
In vivo thrombus formation
Thrombosis was measured in mouse cremaster arterioles as described previously[Citation19]. Briefly, under general anesthesia the cremaster muscle was exteriorized and connective tissue removed. DyLight® 649-conjugated anti-GPIbβ antibody (0.2 µg/g mouse weight) was introduced into the carotid artery via a cannula. Injury to the vessel wall was made with a MicroPoint ablation laser (Andor Technology, Belfast, UK) and thrombus formation recorded using a digital camera with a charge-coupled device (C9300, Hamamatsu Photonics, Welwyn Garden City, UK). Data were analyzed using SlideBook 6 software (Intelligent Imaging Innovations, Denver, USA).
Platelet survival
To determine platelet lifespan, 500 µg of biotin (EZ-link Sulfo NHS-SS-biotin, Thermo Scientific, Paisley, UK) was injected into the tail vein on Day 1. On each subsequent day, 50 µl of blood was collected by tail bleed into ACD. Following centrifugation at 125 g for 10 minutes, PRP was incubated with 20 µl streptavidin-APC (BD Biosciences) for 40 minutes at room temperature in the dark. The sample was washed with NPS and centrifuged for 6 minutes at 860 g, and the platelet pellet resuspended in 0.2% formylsaline for analysis by flow cytometry.
Statistical analysis
All data and statistical analysis were performed using GraphPad Prism 6 (GraphPad Software, Inc, California, USA). Data are presented as mean ± SEM. For parametric data, comparison between 2 groups was performed using the Student t test and significance indicated as not significant (ns), *P ≤ .05, **P ≤ .01 and ***P ≤ .001.
Results
Expression of platelet surface glycoproteins
Deletion of Kv1.3 had no effect on expression of platelet surface glycoproteins GPIbα, GPIbβ, GPV, and integrin subunits α2, β1 and β3 (). In contrast, integrin αIIb was expressed at higher levels on platelets from Kv1.3−/- compared to WT mice (P < .001). Since the αIIb chain forms part of the αIIbβ3 integrin complex, the main receptor for fibrinogen, we tested for possible differences in functional responses in which this major platelet adhesion ligand plays a key role, including aggregation, secretion and thrombus formation.
Table I. Platelet surface glycoprotein expression in WT and Kv1.3−/- mice.
Kv1.3−/- platelets exhibit enhanced ADP-evoked aggregation and secretion
Absence of Kv1.3 was shown to promote platelet aggregation and secretion following P2Y receptor activation with ADP, but not stimulation of GPVI with the collagen peptide, CRP-XL. Platelets from Kv1.3−/- mice displayed an elevated peak aggregation level in response to 1 µM ADP (P = .0395; ), and significantly increased secretion of ATP from dense granules at 1 and 10 μM ADP (P = .0193; and P = .0204, respectively; ). In contrast, aggregation and dense granule secretion were not significantly affected by loss of Kv1.3 in response to CRP-XL at either 1 μg/mL or 10 μg/mL ().
Adhesion to fibrinogen and collagen under flow conditions
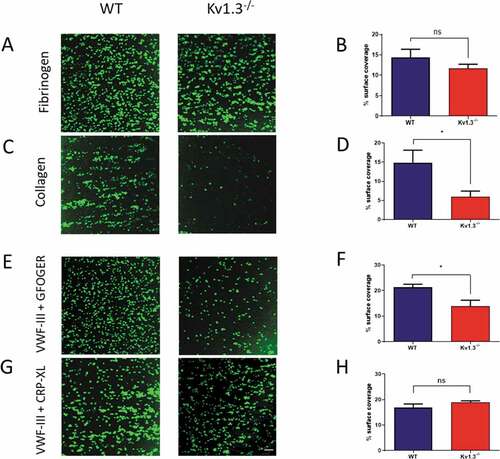
Platelet adhesion to immobilized fibrinogen was not significantly different following perfusion of blood extracted from Kv1.3−/- compared to WT mice (P = .1209; ). In contrast, flow-dependent platelet adhesion to fibrillar collagen was reduced in Kv1.3-deficient platelets (P = .0154; ). Platelet adhesion to collagen under conditions of high/elevated shear is dependent initially on the transient engagement of the platelet GPIb complex with immobilized VWF, which facilitates subsequent direct interaction of platelet collagen receptors α2β1 and GPVI with the collagen fibrils, enabling firm adhesion to take place [Citation20,Citation21]. Under conditions of shear in vitro, platelets perfused over surfaces coated with VWF alone exhibit a rolling across the surface with only brief transient attachment[Citation22]. Therefore we used the triple-helical collagen mimetic peptide, VWF-III, which contains the VWF-A3 binding motif that binds to collagen III, in combination with either the α2β1–specific peptide, GFOGER, or the GPVI-specific peptide, CRP-XL to investigate the contribution of Kv1.3 to platelet adhesion via the two collagen receptors under flow conditions. Platelets from Kv1.3−/- mice had lower surface coverage on coverslips coated with VWF-III + GFOGER compared to platelets from WT mice (P = .0239; ). In contrast, platelet surface coverage on coverslips coated with VWF-III + CRP-XL was not significantly different between the two genotypes (P = .2235; ).
Figure 2. Absence of Kv1.3 reduces integrin α2β1-dependent platelet adhesion to collagen. DiOC6-labeled platelets in whole blood from WT or Kv1.3−/- mice were perfused over fibrinogen (200 µg/mL) at a shear rate of 800 s−1 and collagen (100 µg/mL) at 1800 s−1. After 3 minutes of perfusion the coverslips were washed with normal platelet saline, and the images recorded and quantified as described in ‘Methods.’ Representative images (top panel) show platelet adhesion to fibrinogen (Figure 2A) and collagen (Figure 2C). Statistical analysis shows the percent of platelet surface adhesion (mean ± SEM) on fibrinogen (Figure 2B) and collagen (Figure 2D); (n = 5 for fibrinogen and 4 for collagen). DiOC6-labeled platelets in whole blood from WT or Kv1.3−/- mice were also perfused at a shear rate of 1800 s−1 over coverslips coated with synthetic triple-helical peptides specific for the platelet collagen receptors integrin α2β1 (GFOGER, 100 µg/mL) and GPVI (CRP-XL, 100 µg/mL). Representative images (lower panel) show platelet adhesion to (E) peptides VWF-III and GFOGER, and (G) peptides VWF-III and CRP-XL. Scale bar = 20 µm. Statistical analysis shows the mean percent of platelet surface adhesion to (F) VWF-III and GFOGER (n = 5), and (H) VWF-III and CRP-XL (n = 5). (WT, blue; Kv1.3−/-, red). *P < .05, ns = not significant.
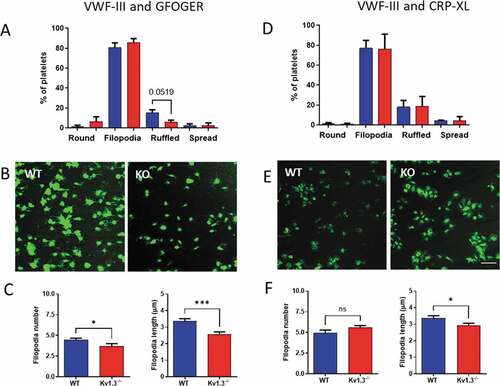
Morphology of platelets adhering to collagen peptides
Adherent platelets were classified into four morphological categories representing different stages of platelet adhesion and activation[Citation23]. Platelets that were rounded and without any protrusions, were classified as ‘round.’ Following initial adhesion, platelets begin to form protrusions to secure adhesion to the collagen fibrils, and these platelets were classified as ‘filopodia.’ Eventually, cytoskeletal remodeling allows actin fibers to fill between the filopodia, and platelets in this category were labeled as ‘ruffled’; and finally, depending on the matrix surface, platelets spread forming the typical ‘fried egg’ appearance.
Following perfusion over the collagen peptides VWF-III and GFOGER a trend toward fewer Kv1.3−/- platelets reaching the ruffled stage was observed, although this failed to reach significance (P = .0519; ), and there was no difference in the percentage of platelets at each stage of platelet adhesion following perfusion over VWF-III and CRP-XL (). Interestingly, despite a similar number of platelets extending filopodia for the two genotypes, deletion of Kv1.3 resulted in a significant reduction in the number of filopodia (P = .0377) and also filopodia length (P = .003) for platelets adhered to VWF-III and GFOGER (). Representative images for platelets attached to VWF-III and GFOGER are shown in and for VWF-III and CRP-XL in .
Figure 3. Kv1.3-deficient platelets form fewer and shorter filopodia during integrin α2β1-dependent adhesion. Platelet morphology following perfusion over collagen peptides at a shear rate of 1800 s−1, was classified as ‘round’ (round platelet with no protrusions), ‘filopodia’ (protrusions securing collagen fibrils), ‘ruffled’ (lamellipodia formation), and ‘spread’ (flattened or fried egg appearance). Distribution of platelet morphology following perfusion of WT and Kv1.3−/- platelets over (A) VWF-III and GFOGER (100 µg/mL each peptide), and (D) VWF-III and CRP-XL (100 µg/mL each peptide). Data is expressed as the mean percent of platelets in each category (n = 5). Representative images of WT and Kv1.3−/- platelet morphology on each peptide surface (B) VWF-III and GFOGER, and (E) VWF-III and CRP-XL. Scale bar = 10 µm. (C, F) Data shows the filopodia number per platelet (based on 50 platelets per treatment group), and filopodia length (µm)(based on length of 100 filopodia per treatment group) of WT and Kv1.3−/-platelets adhered to (C) VWF-III and GFOGER, and (F) VWF-III and CRP-XL. *P < .05, ***P < .005.
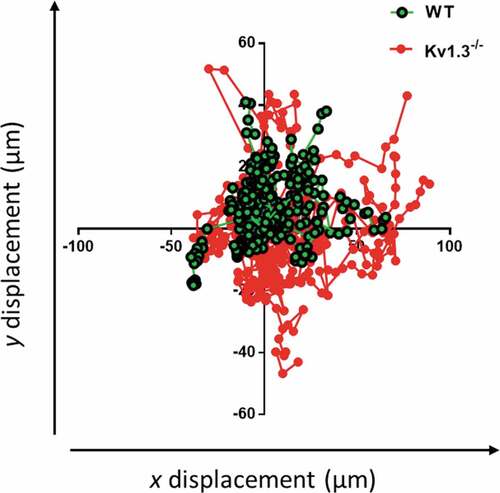
Motile responses of platelets adhering to collagen
To further assess the effect of the altered morphology of Kv1.3−/- platelets during integrin α2β1-dependent platelet adhesion, the motile responses of WT and Kv1.3−/- platelets were tracked during adhesion to fibrillar collagen under static conditions. The tracked trajectories of individual Kv1.3−/- platelets traveled over a wider area than WT platelets (). Observation of video recordings of WT and Kv1.3−/- platelet movement supported these findings of an altered Kv1.3−/- platelet motile response to collagen. During initial attachment to collagen, platelets are able to extend long filopodia toward the collagen fibrils (Supplemental video 1). We observed that WT platelets (Supplemental video 2), rapidly extrude filopodia and pull on the collagen fibers to firmly adhere. In contrast, Kv1.3-deficient platelets displayed a loss of directional persistence, with fewer long filopodia and reduced pulling on collagen fibers (Supplemental video 3).
Figure 4. Kv1.3-deficient platelets lack directional persistence during adhesion to collagen via α2β1. The morphology and motile responses of murine platelets labelled with FM®1-43 lipophilic styryl dye (5 µM) were tracked during adhesion to collagen (100 µg/mL) under static conditions. The plotting of co-ordinates tracked the trajectories of WT and Kv1.3-deficient platelets during platelet attachment to the collagen fibers. Platelet movement and direction is measured by displacement (µm) along the x and y axis. Data from 3 independent experiments.
In vitro thrombus formation on collagen
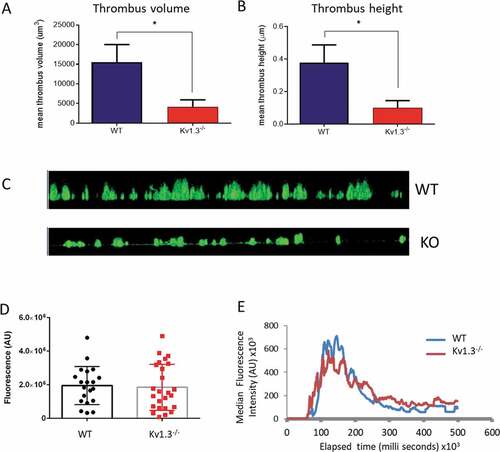
The above experiments demonstrate that deletion of platelet Kv1.3 results in diminished responses involving integrin α2β1, such as adhesion to collagen, but that αIIbβ3-dependent functions, such as aggregation, are not reduced. This suggests that Kv1.3−/- platelets should be able to aggregate and form thrombi where platelet attachment to collagen has been successful. Following platelet perfusion over collagen at a shear rate of 1800 s−1, Kv1.3-deficient platelets that did successfully adhere to collagen fibrils were able to form thrombi, but with significantly reduced height (P = .0254; ), and volume (P = .0254; ). The difference between thrombi formed by WT and Kv1.3−/- platelets on fibrillar collagen can also be seen in the side elevation of z-stack fluorescent images in .
Figure 5. Absence of Kv1.3 reduces in vitro, but not in vivo, thrombus formation. DiOC6-loaded platelets from Kv1.3−/- mice were perfused over collagen-coated coverslips (100 µg/mL), and analyzed for (A) total thrombus volume (µm3) and (B) thrombus height (µm). Data shown is the mean ± SEM for thrombi formed by platelets from WT (blue) and Kv1.3−/- mice (red) (n = 4; *P < .05. (C) Representative side elevation of z-stack fluorescent images of thrombi formed on fibrillar collagen by platelets from WT (upper image) and Kv1.3−/- (lower image) mice. (D) Mean integrated fluorescence (arbitrary units), and (E) Plot of median fluorescence intensity (arbitrary units) over time (seconds), during in vivo thrombus formation following laser-induced injury in cremaster muscle arterioles of WT and Kv1.3−/- mice; (n = 20 thrombi in 5 WT mice and 25 thrombi in 5 Kv1.3−/- mice).
Characterization of platelet function in vivo: thrombus formation
To investigate whether the altered in vitro responses of Kv1.3−/- platelets translate into a change in function within the circulation, we studied thombus formation in the cremaster muscle arterioles of anaesthetised male mice using the laser-induced injury model [Citation24–26]. The profile of median thrombus fluorescence (n = 20 thrombi in five WT mice and 25 thrombi in five Kv1.3−/- mice) through the 500 second duration of thrombus growth and regression in Kv1.3-deficient mice was not significantly different from thrombi formed in WT mice (). Consistent with this, maximum thrombus fluorescence was not altered ().
Platelet size and platelet lifespan
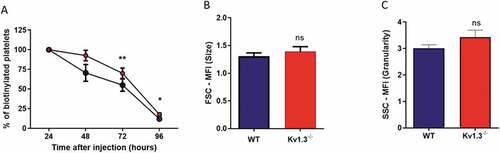
Kv1.3-deficient mice display elevated platelet counts, but no change in megakaryocyte development within the marrow[Citation11]. To explore whether an altered lifespan could account for this phenotype, we measured platelet clearance in Kv1.3−/- and WT mice using an in vivo biotinylation approach[Citation27]. The percent biotinylated platelets was significantly elevated above that measured in the platelet population from WT mice at 72 hours (P = .006) and at 96 hours (P = .0452) post biotin injection (), suggesting that an enhanced lifespan may contribute to the greater platelet count observed in Kv1.3−/- mice. Kv1.3−/- platelets were no different in size (P = .1979; ), or granularity (P = .198; ), as determined by Forward and Side Scatter using flow cytometry.
Figure 6. Platelets from Kv1.3−/- display a longer lifespan. (A) Assessment of platelet lifespan was carried out using in vivo biotinylation of murine platelets, recording the percent of biotinylated platelets isolated from WT and Kv1.3−/- mice over five days (n = 4 for each genotype). Flow cytometric analysis of (B) platelet size and (C) platelet granularity gating on forward scatter and side scatter of platelet populations isolated from WT and Kv1.3−/- mice (n = 8). Data shown is mean ±SEM. **P < .01, *P < .05, ns not significant.
Discussion
Kv1.3 plays a crucial role in maintaining the resting membrane potential in platelets [Citation10,Citation11], regulating entry of the key second messenger Ca2+; however, its contribution to hemostasis and thrombosis is less clear. The present study provides new evidence that loss of Kv1.3 in murine platelets modulates a number of platelet responses, particularly collagen-evoked adhesion and motile responses through a mechanism dependent on integrin α2β1.
Platelet adhesion and thrombus formation relies on two key integrins, namely α2β1 which binds to collagen, and αIIbβ3 which binds several ligands including fibrinogen, fibronectin, Von Willebrand factor (VWF), and fibrin[Citation28]. Our studies of murine platelet adhesion under conditions of arterial shear demonstrate that adhesion to fibrillar collagen, where α2β1 is an important adhesive receptor, is significantly impaired following deletion of Kv1.3. In contrast, loss of Kv1.3 had no effect on αIIbβ3-dependent platelet binding to immobilized fibrinogen. This specific role for Kv1.3 in α2β1-mediated adhesion was further demonstrated in experiments using combinations of triple-helical collagen-specific peptides. Kv1.3−/- platelet adherence was significantly reduced compared to those from WT mice when perfused over surfaces coated with a combination of VWF-III, a peptide which contains the VWF-A3 collagen binding motif, and the integrin α2β1-specific peptide, GFOGER. In contrast, no phenotypic difference was observed between adhesion of WT and Kv1.3−/- platelets perfused over surfaces coated with VWF-III combined with the GPVI-specific collagen peptide, CRP-XL. This is consistent with experiments using human platelets, where Pugh and colleagues used integrin-specific peptides of differing affinity to show that α2β1 enhances the rate of recruitment of platelets to a collagenous surface[Citation29].
Exposure to collagen induces marked changes in platelet morphology. Initial events involve the extension of filopodial protrusions which allow attachment to collagen fibers, followed by formation of actin-rich lamellipodia and eventual transformation to the typical ‘fried egg’ appearance during platelet spreading [Citation23,Citation30]. Mutations that affect the molecular mechanisms and protein interactions of cytosketeletal reorganization during platelet shape change impair the ability of platelets to adhere and form a thrombus [Citation31–34]. Our studies of DiOC6-loaded platelets demonstrate that in the absence of Kv1.3−/-, fewer platelets progress to form lamellipodia (ruffled appearance) on α2β1-dependent surfaces coated with VWF-III/GFOGER; furthermore, Kv1.3−/- platelets exhibited shorter and fewer filopodia compared to WT controls. Video recordings under static conditions show that WT platelets extend long filopodia toward collagen fibers, pulling the fibers upon initial attachment, before spreading (Supplementary video 1 and 2), whereas platelets lacking Kv1.3 are less able to facilitate attachment (Supplementary video 3). Subsequent tracking of the trajectories of individual platelet movement under the same conditions further demonstrated that Kv1.3−/- platelet haptotaxis toward collagen is less efficient, with platelets traveling over a wider area, appearing to have less ability to ‘sense’ the collagen or maintain directional persistence toward it. This is consistent with weaker integrin-dependent adhesion permitting greater motility, as shown in HT1080 fibrosarcoma haptotaxis experiments[Citation35]. Interestingly, blockade or reduced expression of Kv1.3 also impairs migration of T-lymphocytes [Citation36,Citation37], and alters detection of electrical fields in neutrophils[Citation38]. Voltage-gated sodium channels (Nav) have also been proposed to contribute to cellular motility and migration in several types of immune cells including lymphocytes [Citation39] and macrophages [Citation40] and intracellularly localized Nav1.6 supports invasiveness of human breast cancer cells[Citation41]. Sodium channel β subunits are crucial components of the mechanism whereby pore-forming α subunits Nav1.5 or 1.6 regulate adhesion and migration and there is evidence that Navβ1 can act independently as a cell adhesion molecule [Citation42–44].
Thrombus formation by DiOC6-labeled Kv1.3−/- platelets is inhibited during perfusion over collagen-coated surfaces. Our results suggest that this may be due to a Kv1.3-associated contribution to the formation of platelet filopodia and their mechanosensing ability to detect collagen fibrils in the microenvironment, rather than a defect in platelet aggregation. Surprisingly, however, we saw no difference in thrombus formation or thrombus size in cremaster muscle arterioles of WT or Kv1.3−/- mice following laser injury using an in vivo model that causes endothelial damage and collagen exposure[Citation45]. A similar lack of arterial thrombosis phenotype in Kv1.3−/- mice has also recently been reported using alternative models of thrombosis which depend more upon activation by collagen than the laser injury model used in the present study [Citation16]. Therefore, the lack of Kv1.3 iscompensated for in vivo by other pathways. The enhanced aggregation and secretion that we observed in vitro with ADP in Kv1.3−/- platelets may explain this compensation. We observed increased αIIb integrin expression on the surface, however β3 subunit expression was not altered. It is possible that the αIIb subunit is expressed on the surface independently of associated beta subunits; this is known to happen for other integrin subunits [Citation46]. In its monomeric form it would not contribute to enhanced aggregation unless it can combine with other beta subunits, which requires further study. Although Fan and collagues [Citation16] have observed an upregulation in expression of KCNQ4 and other K+ channels in Kv1.3−/-- platelets, we observed no direct evidence for such a change in our previous electrophysiological recordngs [Citation11]. It is known that that secondary activation of P2Y12 receptors by released ADP amplifies collagen-evoked platelet aggregation [Citation47,Citation48]. Thus, the enhanced ADP responses in Kv1.3-deficient platelets may contribute to the lack of significant difference in collagen-evoked aggregation in standard stirred suspension. Given this argument, the reduced thrombus formation under arterial shear is somewhat unexpected. However, the altered motile responses to collagen may be the overriding determinant of whether the platelets initially attach and therefore can generate a thrombus.
We previously reported increased platelet count in Kv1.3−/- mice which was not due to an altered frequency or size of bone marrow MKs[Citation11]. Here we show that significantly higher numbers of biotinylated platelets remained in the circulation of Kv1.3−/- mice post-injection compared to WT. Platelet lifespan in the mouse is around 5 days, and is regulated by components of the intrinsic apoptotic pathway, whereby the pro-survival protein family member, Bcl-xL controls the activity of pro-apoptotic proteins Bak and Bax [Citation49,Citation50]. Studies in lymphocytes have identified Kv1.3 on the inner mitochondrial membrane[Citation51], where it plays a role in the induction of apoptosis through its interaction with Bax [Citation52,Citation53]. Further study is needed to confirm the possible existence and potential contribution of mitoKv1.3 to platelet apoptosis, but it is possible that platelet apoptosis is impaired when Bax cannot interact with mitoKv1.3. This potential role for Kv1.3 in platelet production, and pro-survival phenotype in the absence of Kv1.3, may contribute to elevated levels of platelets in the circulation of the Kv1.3−/- mice. The recent study by Fan and colleagues [Citation16] confirmed the increased platelet count phenotype of Kv1.3−/- mice. without a change in marrow megakaryopoiesis, and additionally demonstrated increased numbers of megakaryocytes in the spleen. This organ is an alternative site of platelet production in the mouse as well as a site of platelet clearance, and thus may also contribute to the increased platelet count following Kv1.3 deletion. The elevated megakaryopoiesis and reduced clearance is likely to increase the number of reticulated platelets in the circulation, which are known to be more reactive than mature platelets [Citation54,Citation55], and this may partially explain the enhanced aggregation and secretory response to low concentrations of ADP in Kv1.3−/- platelets. Although Fan and colleagues [Citation16] concur with a lack of in vivo thrombus formation phenotype of Kv1.3−/- mice, they report that aggregation of platelets to several agonists in vitro, including thrombin and collagen, and high dose ADP (20 µM), are reduced by loss of channel function following either genetic deletion or application of a pore-blocking antibody (6E12#15)[Citation16]. The difference between the two studies requires further investigation but may result from variability in the method of preparing platelets for in vitro studies.
The experiments reported here raise a number of key questions that we have not been able to investigate due to the impact of the COVID-19 pandemic on our laboratories. This challenge has been recognized by Journal editorial policies [Citation56]andwe report here the completed aspects to the work which highlight the need for additional studies. Future experiments should investigate the mechanism responsible for enhanced ADP-evoked aggregation and secretion in Kv1.3−/- platelets and whether responses to other G-protein-coupled receptor agonists are affected. We also propose a pharmacological approach using blockers such as margatoxin and Pap-1. A key area to investigate is the mechanism by which Kv1.3 modulates integrin function and motility; particularly important questions are whether channel opening is required and whether there is an involvement of the K+ channel regulatory proteins identified in our platelet ion channel transcriptome study[Citation12]. The effect of Kv1.3 deletion on interactions with other adhesive substrates is worthwhile investigating, which would benefit from more advanced imaging approaches. Given the enhanced platelet lifespan and increased platelet number in Kv1.3-deficient mice, additional studies should also investigate the presence of mitoKv1.3 and its potential role in the platelet.
Although platelets are highly specialized for hemostasis, they also contribute to immune responses, often serving as a link between the hemostatic and inflammatory systems [Citation57–60]. For example, they facilitate the phagocytic removal and sequestering of pathogens [Citation61–63] and release antimicrobial agents and chemokines [Citation64,Citation65]. Kv1.3 has a well established role in immune function, particularly in T-lymphocytes [Citation66–68] and its overexpression is a common feature of chronic inflammatory diseases, contributing to the over-reaction of cellular immunity and subsequent cytokine storm [Citation69,Citation70]. Interestingly, in a study using the middle cerebral artery occlusion model, a model of ischemic stroke that involves the formation of occlusive platelet thrombi in response to combined thrombotic and inflammatory stimuli[Citation71], the selective Kv1.3 blocker Pap-1 dose-dependently reduced the infarct area in rodents, reducing microglial activation and improving neuronal survival[Citation72]. In light of the present study and work by Fan and colleagues[Citation16], it is worthwhile exploring the relative contribution of platelet Kv1.3 to the etiology of immune disorders.
Key points
The voltage-gated K+ channel Kv1.3 enhances collagen-evoked platelet adhesion and thrombus formation through an α2β1 integrin-dependent mechanism
Kv1.3-deficient platelets display reduced filopodia formation and greater motility during attachment to collagen fibres
Authorship contributions
J.R.W. designed and performed experiments, collected and analsyed data, and wrote the manuscript; S.J. performed and analysed the aggregation and secretion experiments; P.S. performed and analysed the in vivo thrombus studies; L.K.K. kindly provided the Kv1.3−/- mice for the study; I.F. advised on the breeding and genotyping of the mouse colonies and edited the manuscript; R.W.F. provided the collagen peptides and edited the manuscript; J.M.G. supervised the in vivo thrombus experiments and edited the manuscript; and M.P.M-S. designed the study, supervised experiments, interpreted data, and wrote the manuscript.
Supplemental Video 1: FM 1-43-stained wild type platelets attaching to collagen
Download Microsoft Video (AVI) (998.2 KB)Supplemental Video 2: Wild type platelets settling on collagen
Download Microsoft Video (AVI) (3.6 MB)Supplemental Video 3: Kv1.3-deficient platelets settling on collagen
Download Microsoft Video (AVI) (3.3 MB)Acknowledgements
The authors acknowledge the contribution of staff in the Division of Biomedical Services, Preclinical Research Facility, University of Leicester, for technical support and care of experimental animals. We also thank Lory Francescut for excellent technical support, including genotyping. The work was funded by grants from the British Heart Foundation (PG/11/56 and PG/15/21/31355)
Disclosure statement
R.W.F. is Chief Scientific Officer of CambCol Laboratories. Other authors declare no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
Related Research Data
References
- Fadool DA, Tucker K, Perkins R, Fasciani G, Thompson RN, Parsons AD, Overton JM, Koni PA, Flavell RA, Kaczmarek LK, et al. Kv1.3 channel gene-targeted deletion produces “Super-Smeller Mice” with altered glomeruli, interacting scaffolding proteins, and biophysics. Neuron 2004;41(3):451–461. doi:10.1016/S0896-6273(03)00844-4.
- Xu J, Wang P, Li Y, Li G, Kaczmarek LK, Wu Y, Koni PA, Flavell RA, Desir GV. The voltage-gated potassium channel Kv1.3 regulates peripheral insulin sensitivity. Proc Natl Acad Sci U S A 2004;101(9):3112–3117. doi:10.1073/pnas.0308450100.
- Cahalan MD, Chandy KG. The functional network of ion channels in T lymphocytes. Immunol Rev 2009;231(1):59–87.
- Upadhyay SK, Eckel-Mahan KL, Mirbolooki MR, Tjong I, Griffey SM, Schmunk G, Koehne A, Halbout B, Iadonato S, Pedersen B, et al. Selective Kv1.3 channel blocker as therapeutic for obesity and insulin resistance. Proc Natl Acad Sci U S A 2013;110(24):E2239–2248. doi:10.1073/pnas.1221206110.
- Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington MW, Kolski-Andreaco A, Wei E, Grino A, Counts DR, et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci U S A 2006;103(46):17414–17419. doi:10.1073/pnas.0605136103.
- Sarkar S, Nguyen HM, Malovic E, Luo J, Langley M, Palanisamy BN, Singh N, Manne S, Neal M, Gabrielle M, et al. Kv1.3 modulates neuroinflammation and neurodegeneration in Parkinson’s disease. J Clin Invest 2020;130(8):4195–4212. doi:10.1172/JCI136174.
- Leanza L, Henry B, Sassi N, Zoratti M, Chandy KG, Gulbins E, Szabò I. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol Med 2012;4(7):577–593. doi:10.1002/emmm.201200235.
- Teisseyre A, Palko-Labuz A, Sroda-Pomianek K, Michalak K. Voltage-gated potassium channel Kv1.3 as a target in therapy of cancer. Front Oncol 2019;9:933. doi:10.3389/fonc.2019.00933.
- Maruyama Y. A patch-clamp study of mammalian platelets and their voltage-gated potassium current. J Physiol 1987;391:467–485. doi:10.1113/jphysiol.1987.sp016750.
- Mahaut-Smith MP, Rink TJ, Collins SC, Sage SO. Voltage-gated potassium channels and the control of membrane potential in human platelets. J Physiol 1990;428(1):723–735. doi:10.1113/jphysiol.1990.sp018237.
- McCloskey C, Jones S, Amisten S, Snowden RT, Kaczmarek LK, Erlinge D, Goodall AH, Forsythe ID, Mahaut-Smith MP. Kv1.3 is the exclusive voltage-gated K+ channel of platelets and megakaryocytes: roles in membrane potential, Ca2+ signalling and platelet count. J Physiol 2010;588(Pt 9):1399–1406. doi:10.1113/jphysiol.2010.188136.
- Wright JR, Amisten S, Goodall AH, Mahaut-Smith MP. Transcriptomic analysis of the ion channelome of human platelets and megakaryocytic cell lines. Thromb Haemost 2016;116(2):272–284. doi:10.1160/TH15-11-0891.
- Kawa K. Voltage-gated calcium and potassium currents in megakaryocytes dissociated from guinea-pig bone marrow. J Physiol 1990;431(1):187–206. doi:10.1113/jphysiol.1990.sp018326.
- Romero E, Sullivan R. Complexity of the outward K+ current of the rat megakaryocyte. Am J Physiol 1997;272(5 Pt 1):C1525–1531. doi:10.1152/ajpcell.1997.272.5.C1525.
- Kapural L, Feinstein MB, O’Rourke F, Fein A. Suppression of the delayed rectifier type of voltage gated K+ outward current in megakaryocytes from patients with myelogenous leukemias. Blood 1995;86(3):1043–1055. doi:10.1182/blood.V86.3.1043.1043.
- Fan C, Yang X, Wang WW, Wang J, Li W, Guo M, Huang S, Wang Z, Liu K. Role of Kv1.3 channels in platelet functions and thrombus formation. Arterioscler Thromb Vasc Biol 2020;40(10):2360–2375. doi:10.1161/ATVBAHA.120.314278.
- Koni PA, Khanna R, Chang MC, Tang MD, Kaczmarek LK, Schlichter LC, Flavell RA. Compensatory anion currents in Kv1.3 channel-deficient thymocytes. J Biol Chem 2003;278(41):39443–39451. doi:10.1074/jbc.M304879200.
- Pugh N, Simpson AM, Smethurst PA, de Groot PG, Raynal N, Farndale RW. Synergism between platelet collagen receptors defined using receptor-specific collagen-mimetic peptide substrata in flowing blood. Blood 2010;115(24):5069–5079. doi:10.1182/blood-2010-01-260778.
- Sahli K, Flora GD, Sasikumar P, Maghrabi AH, Holbrook LM, AlOuda SK, Elgheznawy A, Sage T, Stainer AR, Adiyaman R, et al. Structural, functional and mechanistic insights uncover the fundamental role of orphan Connexin62 in platelets. Blood 2021;137(6):830-843. doi:10.1182/blood.2019004575.
- Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res 2007;100(12):1673–1685. doi:10.1161/01.RES.0000267878.97021.ab.
- Farndale RW. Collagen-induced platelet activation. Blood Cells Mol Dis 2006;36(2):162–165. doi:10.1016/j.bcmd.2005.12.016.
- McEver RP, Zhu C. Rolling cell adhesion. Annu Rev Cell Dev Biol 2010;26(1):363–396. doi:10.1146/annurev.cellbio.042308.113238.
- Aslan JE, McCarty OJ. Rho GTPases in platelet function. J Thromb Haemost 2013;11(1):35–46. doi:10.1111/jth.12051.
- Rosen ED, Raymond S, Zollman A, Noria F, Sandoval-Cooper M, Shulman A, Merz JL, Castellino FJ. Laser-induced noninvasive vascular injury models in mice generate platelet- and coagulation-dependent thrombi. Am J Pathol 2001;158(5):1613–1622. doi:10.1016/S0002-9440(10)64117-X.
- Celi A, Merrill-Skoloff G, Gross P, Falati S, Sim DS, Flaumenhaft R, Furie BC, Furie B. Thrombus formation: direct real-time observation and digital analysis of thrombus assembly in a living mouse by confocal and widefield intravital microscopy. J Thromb Haemost 2003;1(1):60–68. doi:10.1046/j.1538-7836.2003.t01-1-00033.x.
- Falati S, Gross PL, Merrill-Skoloff G, Sim D, Flaumenhaft R, Celi A, Furie BC, Furie B, In vivo models of platelet function and thrombosis: study of real-time thrombus formation. Methods Mol Biol 2004;272:187–197. doi:10.1385/1-59259-782-3:187.
- Josefsson EC, White MJ, Dowling MR, Kile BT. Platelet life span and apoptosis. Methods Mol Biol 2012;788:59–71.
- Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood 2004;104(6):1606–1615. doi:10.1182/blood-2004-04-1257.
- Pugh N, Bihan D, Perry DJ, Farndale RW. Dynamic analysis of platelet deposition to resolve platelet adhesion receptor activity in whole blood at arterial shear rate. Platelets 2015;26(3):216–219. doi:10.3109/09537104.2014.893289.
- Lickert S, Sorrentino S, Studt JD, Medalia O, Vogel V, Schoen I. Morphometric analysis of spread platelets identifies integrin alphaIIbbeta3-specific contractile phenotype. Sci Rep 2018;8(1):5428. doi:10.1038/s41598-018-23684-w.
- McCarty OJ, Larson MK, Auger JM, Kalia N, Atkinson BT, Pearce AC, Ruf S, Henderson RB, Tybulewicz VLJ, Machesky LM, et al. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem 2005;280(47):39474–39484. doi:10.1074/jbc.M504672200.
- Akbar H, Shang X, Perveen R, Berryman M, Funk K, Johnson JF, Tandon NN, Zheng Y. Gene targeting implicates Cdc42 GTPase in GPVI and non-GPVI mediated platelet filopodia formation, secretion and aggregation. PLoS One 2011;6(7):e22117. doi:10.1371/journal.pone.0022117.
- Barkalow KL, Falet H, Italiano JE Jr., van Vugt A, Carpenter CL, Schreiber AD, Hartwig JH. Role for phosphoinositide 3-kinase in Fc gamma RIIA-induced platelet shape change. Am J Physiol Cell Physiol 2003;285(4):C797–805. doi:10.1152/ajpcell.00165.2003.
- Pleines I, Hagedorn I, Gupta S, May F, Chakarova L, van Hengel J, Offermanns S, Krohne G, Kleinschnitz C, Brakebusch C, et al. Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood 2012;119(4):1054–1063. doi:10.1182/blood-2011-08-372193.
- Farndale RW, Lisman T, Bihan D, Hamaia S, Smerling C, Pugh N, Konitsiotis A, Leitinger B, de Groot P, Jarvis G, et al. Cell-collagen interactions: the use of peptide Toolkits to investigate collagen-receptor interactions. Biochem Soc Trans 2008;36(Pt 2):241–250. doi:10.1042/BST0360241.
- Levite M, Cahalon L, Peretz A, Hershkoviz R, Sobko A, Ariel A, Desai R, Attali B, Lider O. Extracellular K(+) and opening of voltage-gated potassium channels activate T cell integrin function: physical and functional association between Kv1.3 channels and beta1 integrins. J Exp Med 2000;191(7):1167–1176. doi:10.1084/jem.191.7.1167.
- Matheu MP, Beeton C, Garcia A, Chi V, Rangaraju S, Safrina O, Monaghan K, Uemura MI, Li D, Pal S, et al. Imaging of effector memory T cells during a delayed-type hypersensitivity reaction and suppression by Kv1.3 channel block. Immunity 2008;29(4):602–614. doi:10.1016/j.immuni.2008.07.015.
- Kindzelskii AL, Petty HR. Ion channel clustering enhances weak electric field detection by neutrophils: apparent roles of SKF96365-sensitive cation channels and myeloperoxidase trafficking in cellular responses. Eur Biophys J 2005;35(1):1–26. doi:10.1007/s00249-005-0001-2.
- Fraser SP, Diss JK, Lloyd LJ, Pani F, Chioni A-M, George AJT, Djamgoz MBA. T-lymphocyte invasiveness: control by voltage-gated Na+ channel activity. FEBS Lett 2004;569(1–3):191–194. doi:10.1016/j.febslet.2004.05.063.
- Carrithers MD, Chatterjee G, Carrithers LM, Offoha R, Iheagwara U, Rahner C, Graham M, Waxman SG. Regulation of podosome formation in macrophages by a splice variant of the sodium channel SCN8A. J Biol Chem 2009;284(12):8114–8126. doi:10.1074/jbc.M801892200.
- Fraser SP, Diss JK, Chioni AM, Mycielska ME, Pan H, Yamaci RF, Pani F, Siwy Z, Krasowska M, Grzywna Z, et al. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin Cancer Res 2005;11(15):5381–5389. doi:10.1158/1078-0432.CCR-05-0327.
- Brackenbury WJ, Calhoun JD, Chen C, Miyazaki H, Nukina N, Oyama F, Ranscht B, Isom LL. Functional reciprocity between Na+ channel Nav1.6 and β1 subunits in the coordinated regulation of excitability and neurite outgrowth. Proc Natl Acad Sci U S A 2010;107(5):2283–2288. doi:10.1073/pnas.0909434107.
- Chioni AM, Brackenbury WJ, Calhoun JD, Isom LL, Djamgoz MB. A novel adhesion molecule in human breast cancer cells: voltage-gated Na+ channel beta1 subunit. Int J Biochem Cell Biol 2009;41(5):1216–1227. doi:10.1016/j.biocel.2008.11.001.
- Davis TH, Chen C, Isom LL. Sodium channel beta1 subunits promote neurite outgrowth in cerebellar granule neurons. J Biol Chem 2004;279(49):51424–51432. doi:10.1074/jbc.M410830200.
- Sasikumar P, AlOuda KS, Kaiser WJ, Holbrook LM, Kriek N, Unsworth AJ, Bye AP, Sage T, Ushioda R, Nagata K, et al. The chaperone protein HSP47: a platelet collagen binding protein that contributes to thrombosis and hemostasis. J Thromb Haemost 2018;16(5):946–959. doi:10.1111/jth.13998.
- Solovjov DA, Pluskota E, Plow EF. Distinct roles for the alpha and beta subunits in the functions of integrin alphaMbeta2. J Biol Chem 2005;280(2):1336–1345. doi:10.1074/jbc.M406968200.
- Watson SP. Platelet activation by extracellular matrix proteins in haemostasis and thrombosis. Curr Pharm Des 2009;15(12):1358–1372. doi:10.2174/138161209787846702.
- Hechler B, Gachet C. P2 receptors and platelet function. Purinergic Signal 2011;7(3):293–303. doi:10.1007/s11302-011-9247-6.
- Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, et al. Programmed anuclear cell death delimits platelet life span. Cell 2007;128(6):1173–1186. doi:10.1016/j.cell.2007.01.037.
- Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, Iciek LA, Morgan SJ, Nasarre MC, Nelson R, et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ 2007;14(5):943–951. doi:10.1038/sj.cdd.4402081.
- Szabo I, Bock J, Jekle A, Soddemann M, Adams C, Lang F, Zoratti M, Gulbins E. A novel potassium channel in lymphocyte mitochondria. J Biol Chem 2005;280(13):12790–12798. doi:10.1074/jbc.M413548200.
- Szabo I, Bock J, Grassme H, Soddemann M, Wilker B, Lang F, Zoratti M, Gulbins E. Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc Natl Acad Sci U S A 2008;105(39):14861–14866. doi:10.1073/pnas.0804236105.
- Gulbins E, Sassi N, Grassme H, Zoratti M, Szabo I. Role of Kv1.3 mitochondrial potassium channel in apoptotic signalling in lymphocytes. Biochim Biophys Acta 2010;1797(6–7):1251–1259. doi:10.1016/j.bbabio.2010.01.018.
- Ault KA, Rinder HM, Mitchell J, Carmody MB, Vary CP, Hillman RS. The significance of platelets with increased RNA content (reticulated platelets). A measure of the rate of thrombopoiesis. Am J Clin Pathol 1992;98(6):637–646. doi:10.1093/ajcp/98.6.637.
- Lakkis N, Dokainish H, Abuzahra M, Tsyboulev V, Jorgensen J, Ponce De Leon A, Saleem A. Reticulated platelets in acute coronary syndrome: a marker of platelet activity. J Am Coll Cardiol 2004;44(10):2091–2093. doi:10.1016/j.jacc.2004.05.033.
- Watson SP, Harrison P, Halford GM. Editorial policy during the lockdown. Platelets 2020;31(4):411. doi:10.1080/09537104.2020.1758532.
- Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev 2007;21(2):99–111. doi:10.1016/j.blre.2006.06.001.
- von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res 2007;100(1):27–40. doi:10.1161/01.RES.0000252802.25497.b7.
- Vieira-de-Abreu A, Campbell RA, Weyrich AS, Zimmerman GA. Platelets: versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin Immunopathol 2012;34(1):5–30.
- Jenne CN, Urrutia R, Kubes P. Platelets: bridging hemostasis, inflammation, and immunity. Int J Lab Hematol 2013;35(3):254–261. doi:10.1111/ijlh.12084.
- Yeaman MR. The role of platelets in antimicrobial host defense. Clin Infect Dis 1997;25(5):951–968; quiz 969-970. doi:10.1086/516120.
- Fitzgerald JR, Foster TJ, Cox D. The interaction of bacterial pathogens with platelets. Nat Rev Microbiol 2006;4(6):445–457. doi:10.1038/nrmicro1425.
- Gaertner F, Ahmad Z, Rosenberger G, Fan S, Nicolai L, Busch B, Yavuz G, Luckner M, Ishikawa-Ankerhold H, Hennel R, et al. Migrating platelets are mechano-scavengers that collect and bundle bacteria. Cell 2017;171(6):1368–1382 e1323. doi:10.1016/j.cell.2017.11.001.
- Yeaman MR, Bayer AS. Antimicrobial peptides from platelets. Drug Resist Updat 1999;2(2):116–126. doi:10.1054/drup.1999.0069.
- Semple JW, Italiano JE Jr., Freedman J. Platelets and the immune continuum. Nat Rev Immunol 2011;11(4):264–274. doi:10.1038/nri2956.
- DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature 1984;307(5950):465–468. doi:10.1038/307465a0.
- Wulff H, Pennington M. Targeting effector memory T-cells with Kv1.3 blockers. Curr Opin Drug Discov Devel 2007;10(4):438–445.
- Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol 2015;33(1):291–353. doi:10.1146/annurev-immunol-032414-112212.
- Kazama I, Tamada T, Tachi M. Usefulness of targeting lymphocyte Kv1.3-channels in the treatment of respiratory diseases. Inflamm Res 2015;64(10):753–765. doi:10.1007/s00011-015-0855-4.
- Chandy KG, Norton RS. Peptide blockers of Kv1.3 channels in T cells as therapeutics for autoimmune disease. Curr Opin Chem Biol 2017;38:97–107. doi:10.1016/j.cbpa.2017.02.015.
- Braeuninger S, Kleinschnitz C, Nieswandt B, Stoll G. Focal cerebral ischemia. Methods Mol Biol 2012;788:29–42.
- Chen YJ, Nguyen HM, Maezawa I, Jin LW, Wulff H. Inhibition of the potassium channel Kv1.3 reduces infarction and inflammation in ischemic stroke. Ann Clin Transl Neurol 2018;5(2):147–161. doi:10.1002/acn3.513.