
Abstract
Coronavirus disease 2019 (COVID-19) is a pandemic syndrome caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. SARS-CoV-2 infection induces a process of inflammation and thrombosis supported by an altered platelet activation state. This platelet activation is peculiar being characterized by the formation of platelet-leukocytes rather than platelet–platelet aggregates and by an increased procoagulant potential supported by elevated levels of TF positive platelets and microvesicles.
Therapeutic strategies targeting, beyond systemic inflammation (i.e. with tocilizumab, an anti interleukin-6 receptor), this state of platelet activation might therefore be beneficial. Among the antithrombotic drugs proposed as candidates to treat patients with SARS-CoV-2 infection, antiplatelet drugs, such as aspirin are showing promising results.
Introduction
In December 2019, a new infectious disease outbreaked in China causing severe acute respiratory syndrome. The causative agent was identified as a new coronavirus, the SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) [Citation1]. The COVID-19 (Coronavirus Disease 2019) pandemic has led to high morbidity and mortality worldwide challenging healthcare systems and making crucial the identification not only of effective treatments but also of the pathogenic mechanisms.
Clinical features that characterize SARS-CoV-2 infection include inflammation and thrombosis [Citation2,Citation3]. Studies carried out so far have shown high levels of proinflammatory cytokines, the so-called ”cytokine storm” that correlates with disease severity [Citation4–6]. This exacerbated inflammatory state leads to the activation of coagulation, documented by levels of D-Dimer significantly elevated and predictive of mortality [Citation7,Citation8]. Additionally, levels of ferritin, factor VIII, von Willebrand Factor (VWF) and protein of the complement cascade are abnormal in COVID-19 [Citation9–11]. These consistently reported hemostatic changes are indicative for a coagulopathy that may predispose COVID-19 patients to thrombotic events. Postmortem studies have indeed highlighted the presence of multiorgan thrombosis, even in patients on standard thromboprophylaxis [Citation12,Citation13]. Fibrin thrombi in the small arterial vessels were observed in 87% of the analyzed samples. Of note, pulmonary thrombosis was reported in 15% of COVID-19 patients. This incidence, which is similar to that reported in patients with SARS, is more than twofold higher than that reported in patients with H1N1 influenza (6% of patients) [Citation14]. These vascular microthrombi are particularly present in areas of diffuse alveolar damage and support the hypothesis that COVID-19 is complicated by coagulopathy and thrombosis [Citation15].
In the context of this pandemic, platelets can be the potential pathophysiologic drivers of disease associated coagulopathy and thrombotic complications. The critical role of platelets during infections is well known [Citation16]. It has been reported that several viruses, including influenza A virus, coronavirus, dengue virus human immunodeficiency virus type-1 (HIV-1) and adenovirus, are able to bind and activate platelets [Citation17]. On the one side, however, the role of platelets may be beneficial, as they can reduce the circulating viral load by engulfing viruses and presenting them to neutrophils, as has been demonstrated for the influenza A virus [Citation18]. On the other side, activated platelets can support the inflammatory process and tissue injury by worsening the pathological scenario. These cells may also foster the coagulation process by providing both tissue factor, the key protein for the initiation of the coagulation cascade, and the procoagulant surface for the assembly of coagulation factors and trigger the thrombotic process [Citation19,Citation20]. In addition, platelets can amplify inflammation through the release of soluble mediators from intracellular granules, the production of microvesicles and the interaction with leukocytes [Citation21,Citation22]. As a proof of these assumptions, an increased deposition of platelets and megakaryocytes [Citation23,Citation24] as well as an increased number of platelet-leukocyte aggregates has been reported within lung capillaries [Citation15,Citation25].
In this review, we will focus on the platelet alterations associated with COVID-19 disease summarizing changes occurring in platelet number, size, and function that recapitulate an overall state of platelet activation. We will also gain insight into the pharmacological strategies to manage platelet activation during COVID-19.Citation45
Alteration in platelet number and size
Thrombocytopenia is a hallmark of various infectious diseases including MERS and SARS, pathologies in which it was recorded in 37% and 50% of affected patients, respectively [Citation26,Citation27]. It is therefore not surprising that a reduction, although mild, in platelet count is common feature also in patients with severe COVID-19 infection, with a 5–41% incidence depending on disease severity [Citation28]. Several studies, included in a meta-analysis on 7613 COVID-19 patients, reported a significant relationship between thrombocytopenia and the clinical severity and mortality of COVID-19 [Citation29]. Although platelet counts are often within reference ranges and severe thrombocytopenia rarely occurs, reduction of platelet count was identified as a poor prognostic factor [Citation30]. A platelet count <100*109/L in the last 24 h before death in 60% of patients has been reported [Citation31]. This dynamic change in platelet count seems to be peculiar of SARS-Cov-2 infection being not reported with other coronavirus strains [Citation32].
Although changes in platelet number during the course of disease are multifactorial, in the context of infectious diseases a reduced platelet count may be due to decreased production of platelets in damaged lungs, as already described for SARS [Citation33,Citation34]. Alternatively, an increased platelet clearance may occurs as a consequence of the activation of the immune system that induces an antibody-mediated phagocytic response [Citation35]. Finally, the most likely hypothesis is that thrombocytopenia may be caused by increased consumption of platelets due to microtrombi formation [Citation34]. This hypothesis is strengthened by the evidence that in COVID-19 patients there is a trend toward higher levels of immature reticulated platelets, indicative of active cell production by megakaryocytes in response to the increased platelet consumption [Citation28]. This higher platelet turnover leads to the release of young macrothrombocytes that, as such, may account for the increased mean platelet volume found in COVID-19 patients [Citation36–38].
Features of platelet activation in COVID-19
Platelet activation is a common feature of infection diseases and the degree of platelet activation follows the symptoms’ severity. Several reports have shown that platelets from COVID-19 patients are hyperreactive,Citation119 releasing into the blood both dense and alpha granules’ cargo [Citation38–44]. By measuring platelet adhesion to collagen under flow, Zaid et al. reported that both in severe and non-severe COVID-19 patients platelets are more prone to clotting []. Platelet aggregation is also higher than in healthy subjects especially when suboptimal concentrations of agonist are used. This lower platelet stimulation threshold in COVID-19 patients has been reported also by Manne et al., suggesting that infection primes platelet hyper-reactivity. Interestingly, platelet aggregation is not increased when induced by stronger stimuli [Citation46]. Furthermore, Pulcinelli et al. reported that it is even reduced in critically ill patients []. A similar behavior, together with an increased platelet agglutination – mediated by high levels of von Willebrand factor – has been previously described in patients with dengue virus infection []. This finding may be fitted with data reported in other studies showing that the expression of activated glycoprotein IIbIIIa, the gold standard marker of platelet activation, is not altered [Citation38,Citation46].
Platelets respond to the increased systemic inflammation caused by COVID-19 associated cytokine storm increasing the interaction with leukocytes mainly through P-selectin. In accordance with data reported by other research groups [Citation41,Citation47], we have recently highlighted that in COVID-19 patients the higher levels of P-selectin, compared to healthy subjects, expressed on the platelet surface resulted in an enhanced (about 10-fold) interaction with neutrophils and monocytes. Of note, this increased platelet–leukocyte interaction is a feature common also to other virus infection [Citation48,Citation47]. Interestingly, the number of platelet-leukocyte aggregates were significantly lower in critically ill patients compared to those with less severe infection and their levels were negatively associated with plasma levels of IL-6, CRP, and D-dimer, leading to the speculation of a possible involvement of platelet-leukocyte conjugates in microthrombotic events [Citation38]. This hypothesis is supported by results from postmortem studies demonstrating the presence of platelets, neutrophils, and thrombogenic neutrophil-extracellular traps (NETs), whose formation is sustained by platelet-neutrophil aggregates in the lung of COVID-19 patients [Citation12,Citation49,Citation50].
Platelet–neutrophil interaction may also be responsible for the release of NETs, which are highly prothrombotic and, when dysregulated, able to induce intravascular coagulation. It has been shown indeed that sera from patients with COVID-19 contain highly specific markers for NETosis [Citation51] and that NET formation is associated with thrombotic events in COVID-19 patients [Citation40].
Platelet activation in COVID-19 patients also resulted in an increased release of extracellular vesicles (EVs) that might contribute to the COVID-19 pathogenesis [Citation38,Citation43,Citation44]. In the last years, EVs received great attention as potential biomarkers for several human diseases including infections [Citation52,Citation53]. In the context of COVID-19, platelet EVs are strongly associated with SARS-CoV-2 infection, being higher in SARS-CoV-2 positive patients, regardless of the severity of the disease, compared to negative ones. This association is independent from any confounders making EVs a good diagnostic marker as indicated by ROC curve analysis [Citation54].
Coagulation abnormalities and platelet procoagulant phenotype
Among the pathways and mediators that are dysregulated in COVID-19, coagulation disorders are prominent. The clotting abnormalities include prolonged prothrombin time (PT) and partial thromboplastin time (PTT), increased fibrinogen as well as D-dimer levels that are linked with a higher mortality rate [Citation7,Citation55]. An increased resistance to fibrinolysis has also been observed using viscoelastometric tests reinforcing the procoagulant state described in COVID-19 [Citation56,Citation57]. Patients with COVID-19 experience coagulopathies among which pulmonary thromboembolism [Citation58,Citation59], despite LMWH thromboprophylaxis [Citation60] and a not well-defined intravascular coagulation syndrome, characterized by thrombotic events in the absence of a renal consumption coagulopathy and with an apparent lack of bleeding [Citation61]. In a recently published meta-analysis including 20 studies in COVID-19 patients, Di Minno and collaborators have shown that the prevalence of venous thromboembolism (VTE) is ~30%. This percentage rises to 37% when considering only studies in which systematically the screening for thrombosis was carried out [Citation62]. However, the complex relationship between SARS-CoV-2 infection and hemostatic imbalance observed in COVID-19 patients is not fully explained by the traditional risk factors for VTE and other factors seem to be responsible for the observed coagulopathy [Citation62]. Among these, the procoagulant activity of platelets might have particular relevance. In addition to their role in primary hemostasis, platelets are indeed deeply involved in secondary hemostasis, providing not only the surface for the assembly of coagulation factors but also tissue factor (TF), the key protein required to initiate thrombin generation [Citation19]. TF is a transmembrane protein that binds factor VII/VIIa leading to activation of the extrinsic coagulation pathway. TF-dependent activation of coagulation has been described in several viral infections including Herpes simplex virus [Citation63], human immunodeficiency virus (HIV) [Citation64], dengue [Citation65], or Ebola [Citation66]. Different studies have reported increased TF expression in monocytes [Citation41], endothelial cells [Citation67] and extracellular vesicles [Citation68] also in the context of SARS-CoV-2 infection, leading to the speculation that TF may be involved in the prothrombotic phenotype of this disease. We have provided evidence that in COVID-19 patients the number of TF expressing platelets is more than twice greater than that measured in healthy subjects, being higher in critically ill patients. Of note, the protein is functionally active and able to sustain thrombin generation [Citation38]. Unlike the TF expressing platelets, the number of PS positive platelets in COVID-19 patients was comparable to that measured in healthy donors [Citation15,Citation69]. Consistent with the lack of PS exposure, dual-agonist stimulation of platelets from COVID-19 patients did not result in a significant loss of mitochondrial membrane potential [Citation69]. Of interest, Althaus and collaborators highlighted that COVID-19 patients with diagnosis of thromboembolic complications had significantly higher PS externalization compared with those without thrombotic events. It remains unclear whether apoptosis alone is sufficient to support thromboembolic status in these patients or rather this is dependent on the generalized platelet activation state of SARS-CoV-2 disease [Citation70].
The contribution of the extrinsic pathway to thrombosis in COVID-19 patients is further supported by data showing a significant increase in circulating extracellular vesicle-associated TF activity, which is linked with the severity of disease and mortality [Citation71]. Of note, in COVID-19 patients, platelets together with erythrocytes are the main source of TF-positive MVs [Citation38].
Potential mechanisms involved in platelet alterations in COVID-19
Several factors may contribute to the induction of the platelet hyperreactive state described in COVID-19 ().
Figure 1. Platelet alterations in COVID-19 disease. As the SARS-Cov-2 infection progresses, the uncontrolled overproduction of inflammatory cytokines (1) may contribute to platelet activation resulting in increased exposure of P-selectin and Tissue Factor on the surface of circulating platelets.Platelet hyperreactivity may also be a consequence of the effect of SARS-CoV-2 on megakaryocytes. This results in the production of activated platelets characterized by significant changes in their transcriptome (2). Diabetes or cardiovascular diseases, characterized di per se by a sustained platelet activation state, may determine the presence in circulation of COVID-19 patients, of “primed platelets” more prone to activation (3). In addition, the altered platelet activation can be a direct consequence of the virus activity that, once internalized, can determine a Toll like receptor 7-mediated release of platelet granules (4). Finally, activation of endothelial cells, which is another hallmarks of COVID-19 disease, may result in a NO pathway dysfunction (5) that can promote and sustain further platelet activation. These mechanisms, which are not reciprocally exclusive, are responsible for (a) an increase in circulating procoagulant platelets expressing Tissue Factor, which are therefore able to support thrombin generation, and P-selectin-positive platelets available for the formation of heteroaggregates with monocytes and neutrophils (b). The release of alpha and dense granule cargo as well as the production of extracellular vesicles are further consequences of platelet activation (c,d). The interaction between platelets and neutrophils causes the formation of highly prothrombotic NETs (e) that can support the formation of fibrin-rich microthrombi that have been shown in the lung of COVID-19 patients (f,g).
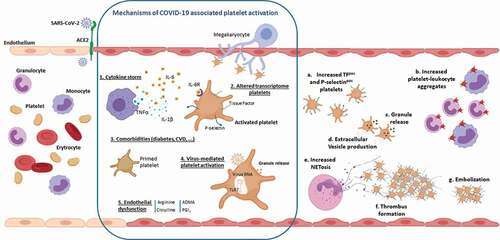
The activation of the immune system, which involves the production of cytokines and immune complements, whereas being essential for the resolution of the infection, can be the cause of immunothrombosis sustained by platelet activation. Circulating platelets can indeed be activated by soluble molecules (e.g. fibrinogen) and inflammatory cytokines, among which IL-6, IL-1b, and TNF-alpha, whose concentrations are significantly elevated in the plasma of COVID-19 patients [Citation72]. Exposure of whole blood to IL-6 and IL-1β can lead to the amplification of platelet activation in response to common agonists, such as ADP or thrombin. In this regard, we have recently characterized the in vitro IL-6-mediated platelet stimulation, in terms of platelet-TF and P-selectin induction as well as of platelet-leukocyte formation. This cell activation, which is prevented by the presence of an IL-6 receptor antagonist, such as tocilizumab, closely mirrors the in vivo platelet activation observed in COVID-19 patients [Citation38].
A second mechanism of platelet activation may lay in an alteration of their transcriptome. Infectious diseases including dengue, influenza, and sepsis are indeed associated with dynamic changes in platelet gene expression [Citation73–75]. Similarly, in COVID-19 virus proliferation within lung tissue may induce activation of megakaryocyte leading to the production of platelets with a significantly altered gene expression profile. Of note, the COVID-19-associated transcriptome differs by hundreds of transcripts from that observed in influenza and sepsis suggesting a unique transcriptional footprint that characterizes SARS-COV-2 infection [Citation46].
Another hypothesis is that numerous COVID-19 comorbidities, typically associated with platelet function alterations, can cause a primed platelet state that worsens COVID-19 illness. Among these, metabolic syndrome, diabetes, gut dysbiosis, cardiovascular disease, cancer, and aging are all conditions known to alter platelet functions [Citation76–78].
Although SARS-CoV-2 RNA in blood is undetectable [Citation38] or present at very low levels in less than 13% of patients with non-severe or severe COVID-19 [Citation79,Citation80], the possibility that the virus can directly activates platelets and/or megakaryocytes cannot be excluded. Indeed, this mechanism has already been described for dengue virus [Citation81], influenza virus [Citation48,Citation82], human immunodeficiency virus-1 (HIV-1) [Citation83], and encephalomyocarditis virus [Citation73].
A putative receptor for the binding and entry of SARS-CoV-2 virus to cells is angiotensin converting enzyme-2 (ACE2) which is also expressed at high levels in the endothelial cells of the lung [Citation84]. Many studies, however, did not detect ACE2 mRNA nor protein in platelets [Citation43,Citation46] not fully confirming the hypothesis that ACE2 is involved in this phenomenon. CD147 may be an alternative receptor for SARS-CoV-2 [Citation85] as already described for SARS-CoV, HIV-1, and measles [Citation86–88]. Although its involvement has been questioned [Citation89], CD 147 is expressed in different cell types including leukocytes, endothelial cells, and platelets [Citation90]. Within blood cells, its expression is higher in males than in females, in line with the prevalence of COVID-19 infection in male, and it is upregulated in asthma, chronic obstructive pulmonary disease, and obesity, that are reported risk factors underlying complications in COVID-19 [Citation91]. Another receptor that might play a role in this presumed mechanism is furin. This convertase is implicated in the activation of the spike S-protein of SARS-CoV-2 [Citation92] and is stored in large amount in platelets [Citation93]. Langnau et al. have recently provided the evidence that plasma levels of furin are associated with poor clinical outcome in COVID-19 patients with cardiovascular diseases. The Authors did not provide the evidence that platelet-derived furin is implicated in SARS-CoV-2 activation, but they speculate that limiting its release by inhibiting platelet activation with antithrombotic drugs may be an effective strategy to prevent worst outcome [Citation93].
Once internalized within platelets, the single strand viral RNA can interact with the Toll-like receptor 7 (TLR7) in the lysosome. Activation of TLR7 may then lead to the release of alpha granules with exposure of P-selectin and CD40L, which mediate interaction with leukocytes. In addition, TLR7 may also lead to the release of complement C3, which promotes the process of neutrophil NETosis [Citation94,Citation95].
Endothelial dysfunction, extensively described in COVID-19, might also be involved in the observed platelet changes. Endothelium synthesis of nitric oxide (NO) and prostaglandin I2, two major molecules controlling platelet activation, is indeed deeply affected in COVID-19 patients. In these subjects, a marked reduction of arginine, which is involved in NO production and whose metabolism is modified by several viral infections [Citation96], as well as a significant increase of ADMA, a specific NOS inhibitor, has also been reported [Citation38]. The endothelial activation in COVID-19 patients is also documented by the significantly increased levels of prostacyclin that, as previously reported in other clinical settings, reflects the homeostatic response to inflammation and platelet activation [Citation97].
Finally, as already described in influenza [Citation98], it may also be hypothesized that, as the infection progresses and the adaptive immune response develops, platelets become further activated as a consequence of the interaction between platelet FcγRIIa and immunoglobulin G antibodies [Citation99].
Possible pharmacological approaches
Despite today several different therapeutic strategies are used to treat SARS-CoV-2 infection, there is not yet a clear consensus on the pharmacological treatment of this disease (COVID-19 Treatment Guidelines Panel, NIH. Available at https://www.covid19treatmentguidelines.nih.gov/). To rapidly identify successful therapies to reduce COVID-19 mortality and morbidity, particular attention has been paid to the repurposing of existing drugs. The current therapeutic strategies are aimed at limiting the dysfunctional processes detected in COVID-19. Antiviral drugs are obviously used to inhibit replication of SARS-CoV-2. Immunomodulatory and anti-inflammatory treatments have proven effective in inhibiting the altered host immune response that leads to the exaggerated release of pro-inflammatory cytokines. Data summarized in two systematic reviews suggest that corticosteroid use in patients with severe COVID-19 disease is associated with reduced mortality [Citation100,Citation101]. Similarly, administration of tocilizumab, an anti-interleukin-6 receptor monoclonal antibody, reduced mortality, and promoted clinical improvement in patients with high concentration of circulating IL-6 [Citation102]. Finally, evidence that the pathobiology of COVID-19 involves thrombosis has led to consideration of the possible efficacy of antiplatelet therapy.
Aspirin and P2Y12 antagonists
In addition to the well-established anti-inflammatory and antithrombotic properties [Citation103], aspirin is effective in reducing replication, propagation, and infectivity of several DNA and RNA viruses, including different human coronavirus (such as the human CoV-229E and the MERS-CoV) [Citation104,Citation105].
Additionally, aspirin is able to reduce NETs’ release in a sepsis model, thus limiting their potential to induce thrombin generation and drive intravascular coagulation [Citation106]. Data in the literature reported that chronic intake of aspirin (100 mg/day) was effective in reducing mortality rate in patients with community-onset pneumonia compared to those not receiving the antiplatelet treatment (HR 2.07; 95% CI; 1.08-.3.98; p = .029; for all-cause mortality) [Citation107].
A recent meta-analysis of 10 cohort studies enrolling more than 680,000 patients with sepsis indicated that aspirin, administered before and after the onset of sepsis, reduced the rate of admission to ICU and of hospital mortality [Citation108]. Aspirin is also effective in reducing the incidence of ARDS (OD 0.59, 95% CI, 0.36–0.98) [Citation109]. Even clopidogrel administration, in association with an antiviral agent, improved survival rate during influenza infection in mice [Citation47].
Altogether, these evidences provide the rationale for hypothesizing a benefit of pharmacological treatment of SARS-CoV-2 infection with these drugs. Thus, targeting both platelet activation as well as virus replication, antiplatelet drugs can be an effective therapeutic option.
To date, there are few observational studies reporting the effect of antiplatelet treatment with aspirin and/or antiP2Y12 on COVID-19. In a very recent retrospective study, Chow et al. examined the association between aspirin use and the clinical outcome in 412 hospital-admitted COVID-19 patients. Even though the patients treated with the antiplatelet drug had higher rates of cardiovascular risk factors and coronary artery disease, aspirin treatment was independently associated with a reduced risk of mechanical ventilation, intensive care unit admission, as well as in-hospital mortality [Citation110]. Accordingly, Liu and collaborators showed that low-dose aspirin treatment (100 mg/day) among hospitalized COVID-19 patients was associated with lower risk of mortality compared with a matched group of non-aspirin users [Citation111]. Viecca et al. showed that the combined use of several antiplatelet therapies in severe COVID-19 patients with a thrombophilic profile improved gas exchange efficiency and increased arterial oxygenation [Citation112]. Finally, we have provided the evidence that in vitro aspirin and the treatment with a P2Y12 inhibitor prevent platelet activation induced by plasma of COVID-19 patients [Citation38]. The evidence that inhibition of P2Y12 affects the release of sIL-6 R, the specific soluble receptor needed for IL-6 provides another possible mechanism underpinning the clinical efficacy observed in the treatment of COVID-19 [Citation113].
Other pleiotropic effects can also be involved in the effectiveness of aspirin treatment, such as its effects on endothelial function. Aspirin increases NO availability by acetylating endothelial NO synthase (eNOS), by increasing the activity of dimethylarginine-dimethylaminohydrolase, which is responsible for the degradation of ADMA and by promoting the generation of 15-epi lipoxin A4, which increases eNOS activity [Citation114,Citation115].
Finally, results obtained in a retrospective population‐based cross‐sectional study by Merzon and collaborators have recently suggested that preexisting treatment with low-dose aspirin might have a protective effect on COVID-19 susceptibility and disease duration among COVID-19-infected subjects [Citation116]. This finding, if confirmed by appropriate trials, would even pave the way to the use of low-doses of aspirin for the prevention of COVID-19 infection.
The effectiveness of antithrombotic therapy certainly requires further investigation. Results of the RECOVERY trial have recently become available (currently unpublished and under peer-review) [Citation117]. The study included more than 15000 patients randomized to receive aspirin or usual care alone. The data suggest that aspirin use is associated with an absolute reduction in thrombotic events (4.6% vs 5.3%) although it is not associated with a reduction in 28-day mortality. It should be mentioned, however, that, as the Authors stated, the rate of thromboembolic events in the studied population was lower compared to previous reports [Citation118,119]. It is plausible therefore that aspirin might have more meaningful benefit in a higher thrombotic risk population. Several other randomized clinical trials are, however, currently in progress to clarify whether the use of antiplatelet drugs, such as aspirin (ClinicalTrials.gov Identifier: NCT04324463, NCT04365309, NCT04324463, NCT04363840) or clopidogrel (ClinicalTrials.gov Identifier: NCT02735707, NCT04409834; NCT04333407), could be potentially useful to mitigate the clinical consequence of SARS-CoV-2 infection.
Conclusion
In summary, during the acute phase of SARS-Cov-2 infection, platelets are characterized by an activated phenotype with enhanced TF expression, and release of extracellular vesicles together with a pronounced increase in the number of platelet-leukocyte aggregates. Activated platelets interact with neutrophils, promoting the process of NETosis, and with a dysfunctional endothelium inducing a prothrombotic phenotype. As the infection progresses, the cytokine storm first, and the interaction with antibody immunoglobulins later on can amplify platelet activation ultimately responsible for the prothrombotic scenario characterizing SARS-CoV-2 infection.
Data available on the effectiveness of aspirin and anti P2Y12 provide a strong rationale for proposing the use of antiplatelet drugs in the treatment of COVID-19.
Declaration of interest
No potential competing interest was reported by the authors.
References
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020; 3828: 727–733. https://doi.org/10.1056/NEJMoa2001017
- Joly BS, Siguret V, Veyradier A. Understanding pathophysiology of hemostasis disorders in critically ill patients with COVID-19. Intensive Care Med. 2020. 468: 1603–1606.https://doi.org/10.1007/s00134-020-06088-1
- Klok FA, Kruip M, NJM VDM, Arbous MS, Gommers D, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020; 191: 145–147. https://doi.org/10.1016/j.thromres.2020.04.013
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020; 39510223: 497–506. https://doi.org/10.1016/S0140-6736(20)30183-5
- Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, Jordan TX, Oishi K, Panis M, Sachs D, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020; 1815: 1036–1045 e9. https://doi.org/10.1016/j.cell.2020.04.026
- Sun X, Wang T, Cai D, Hu Z, Chen J, Liao H, Zhi L, Wei H, Zhang Z, Qiu Y, et al. Cytokine storm intervention in the early stages of COVID-19 pneumonia. Cytokine Growth Factor Rev. 2020; 53: 38–42. https://doi.org/10.1016/j.cytogfr.2020.04.002
- Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020; 39510229: 1054–1062. https://doi.org/10.1016/S0140-6736(20)30566-3
- Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020. 184: 844–847.https://doi.org/10.1111/jth.14768
- Henry BM, MHS DO, Benoit S, Plebani M, Lippi G. Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): a meta-analysis. Clin Chem Lab Med. 2020. 587: 1021–1028.https://doi.org/10.1515/cclm-2020-0369
- Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, Baluha A, Bar N, Bona RD, Burns AJ, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020; 78: e575–e582. https://doi.org/10.1016/S2352-3026(20)30216-7
- Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, Baxter-Stoltzfus A, Laurence J Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res 2020;220:1–13.
- Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020; 3832: 120–128. https://doi.org/10.1056/NEJMoa2015432
- Rapkiewicz AV, Mai X, Carsons SE, Pittaluga S, Kleiner DE, Berger JS, Thomas S, Adler NM, Charytan DM, Gasmi B, et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: a case series. EClinicalMedicine. 2020; 24: 100434. https://doi.org/10.1016/j.eclinm.2020.100434
- LP H, CM N, AR S, RA I, JH M, JA V, Vinarsky V, Rubin J, DA O, Sclafani A, et al. Lung histopathology in coronavirus disease 2019 as compared with severe acute respiratory syndrome and h1n1 influenza: a systematic review. Chest. 2021; 1591: 73–84. https://doi.org/10.1016/j.chest.2020.09.259
- Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, Zerbi P, Rech R, Colombo R, Antinori S, Corbellino M, et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis. 2020; 2010: 1135–1140. https://doi.org/10.1016/S1473-3099(20)30434-5
- Portier I, Campbell RA. Role of platelets in detection and regulation of infection. Arterioscler Thromb Vasc Biol. 2021; 41: 70–78
- Antoniak S, Mackman N. Platelets and viruses. Platelets. 2021. 323: 325–330.https://doi.org/10.1080/09537104.2021.1887842
- Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, Yao C, Rade J, Levy D, Wang JP, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. 2019; 101: 1780. https://doi.org/10.1038/s41467-019-09607-x
- Brambilla M, Facchinetti L, Canzano P, Rossetti L, Ferri N, Balduini A, Abbonante V, Boselli D, De Marco L, Di Minno MN, et al. Human megakaryocytes confer tissue factor to a subset of shed platelets to stimulate thrombin generation. Thromb Haemost. 2015; 1149: 579–592. https://doi.org/10.1160/TH14-10-0830
- Dahlback B. Blood coagulation. Lancet. 2000. 3559215: 1627–1632.https://doi.org/10.1016/S0140-6736(00)02225-X
- Margraf A, Zarbock A. Platelets in Inflammation and Resolution. J Immunol. 2019. 2039: 2357–2367.https://doi.org/10.4049/jimmunol.1900899
- Stokes KY, Granger DN. Platelets: a critical link between inflammation and microvascular dysfunction. J Physiol. 2012. 5905: 1023–1034.https://doi.org/10.1113/jphysiol.2011.225417
- McMullen PD, Cho JH, Miller JL, Husain AN, Pytel P, Krausz T, Descriptive A. Quantitative immunohistochemical study demonstrating a spectrum of platelet recruitment patterns across pulmonary infections including COVID-19. Am J Clin Pathol. 2021. 1553: 354–363.https://doi.org/10.1093/ajcp/aqaa230
- Dwiputra Hernugrahanto K, Novembri Utomo D, Hariman H, Budhiparama NC, Medika Hertanto D, Santoso D, Hogendoorn PCW. Thromboembolic involvement and its possible pathogenesis in COVID-19 mortality: lesson from post-mortem reports. Eur Rev Med Pharmacol Sci. 2021; 25: 1670–1679
- Buja LM, Wolf DA, Zhao B, Akkanti B, McDonald M, Lelenwa L, Reilly N, Ottaviani G, Elghetany MT, Trujillo DO, et al. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc Pathol. 2020; 48: 107233. https://doi.org/10.1016/j.carpath.2020.107233
- Hwang SM, Na BJ, Jung Y, Lim HS, Seo JE, Park SA, Cho YS, Song EH, Seo JY, Kim SR, et al. Clinical and laboratory findings of middle east respiratory syndrome coronavirus infection. Jpn J Infect Dis. 2019; 723: 160–167. https://doi.org/10.7883/yoken.JJID.2018.187
- Wong RS, Wu A, To KF, Lee N, Lam CW, Wong CK, Chan PK, Ng MH, Yu LM, Hui DS, Tam JS, Cheng G, Sung JJ. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ. 2003; 3267403: 1358–1362. https://doi.org/10.1136/bmj.326.7403.1358
- Wool GD, Miller JL. The impact of COVID-19 disease on platelets and coagulation. Pathobiology. 2021. 881: 15–27.https://doi.org/10.1159/000512007
- Jiang SQ, Huang QF, Xie WM, Lv C, Quan XQ. The association between severe COVID-19 and low platelet count: evidence from 31 observational studies involving 7613 participants. Br J Haematol. 2020. 1901: e29–e33.https://doi.org/10.1111/bjh.16817
- Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta. 2020; 506: 145–148. https://doi.org/10.1016/j.cca.2020.03.022
- Zhang B, Zhou X, Qiu Y, Song Y, Feng F, Feng J, Song Q, Jia Q, Wang J, Jin X. Clinical characteristics of 82 cases of death from COVID-19. PLoS One. 2020. 157: e0235458.https://doi.org/10.1371/journal.pone.0235458
- Kim JK, Jeon JS, Kim JW, Kim GY. Correlation between abnormal platelet count and respiratory viral infection in patients from Cheonan, Korea. J Clin Lab Anal. 2016. 303: 185–189.https://doi.org/10.1002/jcla.21822
- Ren LL, Wang YM, Wu ZQ, Xiang ZC, Guo L, Xu T, Jiang YZ, Xiong Y, Li YJ, Li XW, et al. Identification of a novel coronavirus causing severe pneumonia in human: a descriptive study. Chin Med J (Engl). 2020; 1339: 1015–1024. https://doi.org/10.1097/CM9.0000000000000722
- Xu P, Zhou Q, Xu J. Mechanism of thrombocytopenia in COVID-19 patients. Ann Hematol. 2020. 996: 1205–1208.https://doi.org/10.1007/s00277-020-04019-0
- Semple JW, Aslam R, Kim M, Speck ER, Freedman J. Platelet-bound lipopolysaccharide enhances Fc receptor-mediated phagocytosis of IgG-opsonized platelets. Blood. 2007. 10911: 4803–4805.https://doi.org/10.1182/blood-2006-12-062695
- Guclu E, Kocayigit H, Okan HD, Erkorkmaz U, Yurumez Y, Yaylaci S, Koroglu M, Uzun C, Karabay O. Effect of COVID-19 on platelet count and its indices. Rev Assoc Med Bras. 1992; 202066: 1122–1127
- Cohen A, Harari E, Cipok M, Laish-Farkash A, Bryk G, Yahud E, Sela Y, Lador NK, Mann T, Mayo A. et al. Immature platelets in patients hospitalized with Covid-19. J Thromb Thrombolysis. 2021 Apr;51(3):608-616.https://doi.org/10.1007/s11239-020-02290-6
- Canzano P, Brambilla M, Porro B, Cosentino N, Tortorici E, Vicini S, Poggio P, Cascella A, Pengo MF, Veglia F, et al. Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. JACC Basic Transl Sci. 2021; 63: 202–218. https://doi.org/10.1016/j.jacbts.2020.12.009
- Comer SP, Cullivan S, Szklanna PB, Weiss L, Cullen S, Kelliher S, Smolenski A, Murphy C, Altaie H, Curran J, et al. COVID-19 induces a hyperactive phenotype in circulating platelets. PLoS Biol. 2021; 192: e3001109. https://doi.org/10.1371/journal.pbio.3001109
- Petito E, Falcinelli E, Paliani U, Cesari E, Vaudo G, Sebastiano M, Cerotto V, Guglielmini G, Gori F, Malvestiti M, et al. Association of neutrophil activation, more than platelet activation, with thrombotic complications in coronavirus disease 2019. J Infect Dis. 2021; 2236: 933–944. https://doi.org/10.1093/infdis/jiaa756
- Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pao CRR, Righy C, Franco S, Souza TML, Kurtz P, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. 2020; 13611: 1330–1341. https://doi.org/10.1182/blood.2020007252
- Barrett TJ, Lee AH, Xia Y, Lin LH, Black M, Cotzia P, Hochman J, Berger JS. Platelet and vascular biomarkers associate with thrombosis and death in coronavirus disease. Circ Res. 2020. 1277: 945–947.https://doi.org/10.1161/CIRCRESAHA.120.317803
- Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, Limami Y, Zaid N, Sadki K, Ben El Haj R, et al. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ Res. 2020; 12711: 1404–1418. https://doi.org/10.1161/CIRCRESAHA.120.317703
- Krishnamachary B, Cook C, Spikes L, Chalise P, Dhillon NK.The potential role of extracellular vesicles in COVID-19 associated endothelial injury and pro-inflammation. medRxiv [Preprint]. 2020 Sep 1:2020.08.27.20182808. https://doi.org/10.1101/2020.08.27.20182808
- Pulavendran S, Rudd JM, Maram P, Thomas PG, Akhilesh R, Malayer JR, Chow VTK, Teluguakula N. Combination therapy targeting platelet activation and virus replication protects mice against lethal influenza pneumonia. American journal of respiratory cell and molecular biology. 2019; 61: 689–701
- Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, Petrey AC, Tolley ND, Guo L, Cody M, et al. Platelet gene expression and function in patients with COVID-19. Blood. 2020; 13611: 1317–1329. https://doi.org/10.1182/blood.2020007214
- Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, Liu M, Zhao X, Xie Y, Yang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. 2020; 131: 120. https://doi.org/10.1186/s13045-020-00954-7
- Rondina MT, Brewster B, Grissom CK, Zimmerman GA, Kastendieck DH, Harris ES, Weyrich AS. In Vivo platelet activation in critically iii patients with primary 2009 influenza A(H1N1). Chest. 2012. 1416: 1490–1495.https://doi.org/10.1378/chest.11-2860
- Dolhnikoff M, Duarte-Neto AN, de Almeida Monteiro RA, LFF DS, de Oliveira EP, Saldiva PHN, Mauad T, Negri EM. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J Thromb Haemost. 2020. 186: 1517–1519.https://doi.org/10.1111/jth.14844
- Rendeiro AF, Ravichandran H, Bram Y, Chandar V, Kim J, Meydan C, Park J, Foox J, Hether T, Warren S, et al. The spatial landscape of lung pathology during COVID-19 progression. Nature. 2021; 5937860: 564–569. https://doi.org/10.1038/s41586-021-03475-6
- Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair C, Weber A, Barnes BJ, Egeblad M, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020 Jun 4;5(11):e138999. https://doi.org/10.1172/jci.insight.138999.
- Urbanelli L, Buratta S, Tancini B, Sagini K, Delo F, Porcellati S, Emiliani C. The role of extracellular vesicles in viral infection and transmission. Vaccines (Basel). 2019 Aug 28;7(3):102. https://doi.org/10.3390/vaccines7030102.
- Camera M, Brambilla M, Canzano P, Cavallotti L, Parolari A, Tedesco CC, Zara C, Rossetti L, Tremoli E. Association of microvesicles with graft patency in patients undergoing CABG surgery. J Am Coll Cardiol. 2020. 7522: 2819–2832.https://doi.org/10.1016/j.jacc.2020.03.073
- Cappellano G, Raineri D, Rolla R, Giordano M, Puricelli C, Vilardo B, Manfredi M, Cantaluppi V, Sainaghi PP, Castello L, et al. Circulating platelet-derived extracellular vesicles are a hallmark of sars-Cov-2 infection. Cells. 2021 Jan 7;10(1):85. https://doi.org/10.3390/cells10010085.
- Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020. 185: 1094–1099.https://doi.org/10.1111/jth.14817
- Panigada M, Bottino N, Tagliabue P, Grasselli G, Novembrino C, Chantarangkul V, Pesenti A, Peyvandi F, Tripodi A. Hypercoagulability of COVID-19 patients in intensive care unit: a report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost. 2020. 187: 1738–1742.https://doi.org/10.1111/jth.14850
- Spiezia L, Boscolo A, Poletto F, Cerruti L, Tiberio I, Campello E, Navalesi P, Simioni P, Cerruti L, Cola M. COVID-19-related severe hypercoagulability in patients admitted to intensive care unit for acute respiratory failure. Thromb Haemost. 2020. 12010: 998–1000.https://doi.org/10.1055/s-0040-1714350
- Danzi GB, Loffi M, Galeazzi G, Gherbesi E. Acute pulmonary embolism and COVID-19 pneumonia: a random association? Eur Heart J. 2020; 4119: 1858
- Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. 2020. 186: 1421–1424.https://doi.org/10.1111/jth.14830
- Khan IH, Savarimuthu S, Leung MST, Harky A. The need to manage the risk of thromboembolism in COVID-19 patients. J Vasc Surg. 2020. 723: 799–804.https://doi.org/10.1016/j.jvs.2020.05.015
- Levi M, Iba T. COVID-19 coagulopathy: is it disseminated intravascular coagulation? Intern Emerg Med. 2021. 162: 309–312.https://doi.org/10.1007/s11739-020-02601-y
- Di Minno A, Ambrosino P, Calcaterra I, Di Minno MNDCOVID-19. and venous thromboembolism: a meta-analysis of literature studies. Semin Thromb Hemost. 2020. 467: 763–771.https://doi.org/10.1055/s-0040-1715456
- Key NS, Vercellotti GM, Winkelmann JC, Moldow CF, Goodman JL, Esmon NL, Esmon CT, Jacob HS. Infection of vascular endothelial cells with herpes simplex virus enhances tissue factor activity and reduces thrombomodulin expression. Proc Natl Acad Sci U S A. 1990. 8718: 7095–7099.https://doi.org/10.1073/pnas.87.18.7095
- Funderburg NT, Mayne E, Sieg SF, Asaad R, Jiang W, Kalinowska M, Luciano AA, Stevens W, Rodriguez B, Brenchley JM, et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood. 2010; 1152: 161–167. https://doi.org/10.1182/blood-2009-03-210179
- Huerta-Zepeda A, Cabello-Gutierrez C, Cime-Castillo J, Monroy-Martinez V, Manjarrez-Zavala ME, Gutierrez-Rodriguez M, Izaguirre R, Ruiz-Ordaz BH. Crosstalk between coagulation and inflammation during dengue virus infection. Thromb Haemost. 2008. 9911: 936–943.https://doi.org/10.1160/TH07-08-0483
- Geisbert TW, Young HA, Jahrling PB, Davis KJ, Kagan E, Hensley LE. Mechanisms underlying coagulation abnormalities in Ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J Infect Dis. 2003. 18811: 1618–1629.https://doi.org/10.1086/379724
- DiNicolantonio JJ, McCarty M. Thrombotic complications of COVID-19 may reflect an upregulation of endothelial tissue factor expression that is contingent on activation of endosomal NADPH oxidase.Open Heart. 2020 Jun;7(1):e001337. https://doi.org/10.1136/openhrt-2020-001337
- Rosell A, Havervall S, von Meijenfeldt F, Hisada Y, Aguilera K, Grover SP, Lisman T, Mackman N, Thalin C. Patients with COVID-19 have elevated levels of circulating extracellular vesicle tissue factor activity that is associated with severity and mortality-brief report. Arterioscler Thromb Vasc Biol. 2021. 412: 878–882.https://doi.org/10.1161/ATVBAHA.120.315547
- Denorme F, Manne BK, Portier I, Petrey AC, Middleton EA, Kile BT, Rondina MT, Campbell RA. COVID-19 patients exhibit reduced procoagulant platelet responses. J Thromb Haemost. 2020. 1811: 3067–3073.https://doi.org/10.1111/jth.15107
- Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Haberle H, Mehrlander M, Hammer S, Schulze H, Bitzer M, et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood. 2021; 1378: 1061–1071. https://doi.org/10.1182/blood.2020008762
- Rosell A, Havervall S, von Meijenfeldt F, Hisada Y, Aguilera K, Grover SP, Lisman T, Mackman N, Thalin C. With Patients COVID-19 have elevated levels of circulating extracellular vesicle tissue factor activity that is associated with severity and mortality. Arterioscler Thromb Vasc Biol. 2021 Feb;41(2):878-882. https://doi.org/10.1161/ATVBAHA.120.315547
- Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science. 2020. 3686490: 473–474.https://doi.org/10.1126/science.abb8925
- Campbell RA, Schwertz H, Hottz ED, Rowley JW, Manne BK, Washington AV, Hunter-Mellado R, Tolley ND, Christensen M, Eustes AS, et al. Human megakaryocytes possess intrinsic antiviral immunity through regulated induction of IFITM3. Blood. 2019; 13319: 2013–2026. https://doi.org/10.1182/blood-2018-09-873984
- Middleton EA, Rowley JW, Campbell RA, Grissom CK, Brown SM, Beesley SJ, Schwertz H, Kosaka Y, Manne BK, Krauel K, et al. Sepsis alters the transcriptional and translational landscape of human and murine platelets. Blood. 2019; 13412: 911–923. https://doi.org/10.1182/blood.2019000067
- Davizon-Castillo P, Rowley JW, Rondina MT. Megakaryocyte and platelet transcriptomics for discoveries in human health and disease. Arterioscler Thromb Vasc Biol. 2020. 406: 1432–1440.https://doi.org/10.1161/ATVBAHA.119.313280
- Zhu L, She ZG, Cheng X, Qin JJ, Zhang XJ, Cai J, Lei F, Wang H, Xie J, Wang W. et al. Association of blood glucose control and outcomes in patients with COVID-19 and pre-existing type 2 diabetes. Cell Metab. 2020; 31: 1068–1077 e3
- Gupta AK, Jneid H, Addison D, Ardehali H, Boehme AK, Borgaonkar S, Boulestreau R, Clerkin K, Delarche N, DeVon HA, et al. Current perspectives on coronavirus disease 2019 and cardiovascular disease: a white paper by the JAHA editors. Journal of the American Heart Association. 2020; 9
- Flook M, Jackson C, Vasileiou E, Simpson CR, Muckian MD, Agrawal U, McCowan C, Jia Y, Murray JLK, Ritchie LD, et al. Informing the public health response to COVID-19: a systematic review of risk factors for disease, severity, and mortality. BMC Infect Dis. 2021; 211: 342. https://doi.org/10.1186/s12879-021-05992-1
- Parra-Izquierdo I, Bradley R, Aslan JE. Platelets get gutted by PAG. Platelets. 2020. 315: 618–620.https://doi.org/10.1080/09537104.2020.1759793
- Andersson MI, Arancibia-Carcamo CV, Auckland K, Baillie JK, Barnes E, Beneke T, Bibi S, Brooks T, Carroll M, Crook D, et al. SARS-CoV-2 RNA detected in blood products from patients with COVID-19 is not associated with infectious virus. Wellcome Open Res. 2020; 5: 181. https://doi.org/10.12688/wellcomeopenres.16002.2
- Vogt MB, Lahon A, Arya RP, Spencer Clinton JL, Rico-Hesse R. Dengue viruses infect human megakaryocytes, with probable clinical consequences. PLoS Negl Trop Dis. 2019 Nov 25;13(11):e0007837. https://doi.org/10.1371/journal.pntd.0007837
- Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, Yao C, Rade J, Levy D, Wang JP, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. 2019 Apr 16;10(1):1780. https://doi.org/10.1038/s41467-019-09607-x.
- Laurence J, Elhadad S, Ahamed J. HIV-associated cardiovascular disease: importance of platelet activation and cardiac fibrosis in the setting of specific antiretroviral therapies. Open Heart. 2018. 52: e000823.https://doi.org/10.1136/openhrt-2018-000823
- Li R, Qiao S, Zhang G. Analysis of angiotensin-converting enzyme 2 (ACE2) from different species sheds some light on cross-species receptor usage of a novel coronavirus 2019-nCoV. J Infection. 2020; 80: 469–472
- Wang K, Chen W, Zhang Z, Deng Y, Lian JQ, Du P, Wei D, Zhang Y, Sun XX, Gong L, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther. 2020; 51: 283. https://doi.org/10.1038/s41392-020-00426-x
- Watanabe A, Yoneda M, Ikeda F, Terao-Muto Y, Sato H, Kai C. CD147/EMMPRIN acts as a functional entry receptor for measles virus on epithelial cells. J Virol. 2010. 849: 4183–4193.https://doi.org/10.1128/JVI.02168-09
- Pushkarsky T, Zybarth G, Dubrovsky L, Yurchenko V, Tang H, Guo H, Toole B, Sherry B, Bukrinsky M. CD147 facilitates HIV-1 infection by interacting with virus-associated cyclophilin A. Proc Natl Acad Sci U S A. 2001. 9811: 6360–6365.https://doi.org/10.1073/pnas.111583198
- Chen Z, Mi L, Xu J, Yu J, Wang X, Jiang J, Xing J, Shang P, Qian A, Li Y, et al. Function of HAb18G/CD147 in invasion of host cells by severe acute respiratory syndrome coronavirus. J Infect Dis. 2005; 1915: 755–760. https://doi.org/10.1086/427811
- Shilts J, Crozier TWM, Greenwood EJD, Lehner PJ, Wright GJ. No evidence for basigin/CD147 as a direct SARS-CoV-2 spike binding receptor. Sci Rep. 2021. 111: 413.https://doi.org/10.1038/s41598-020-80464-1
- Heinzmann D, Noethel M, Ungern-Sternberg SV, Mitroulis I, Gawaz M, Chavakis T, May AE, Seizer P. CD147 is a Novel Interaction Partner of Integrin alphaMbeta2 Mediating Leukocyte and Platelet Adhesion. Biomolecules. 2020 Apr 2;10(4):541. https://doi.org/10.3390/biom10040541.
- Radzikowska U, Ding M, Tan G, Zhakparov D, Peng Y, Wawrzyniak P, Wang M, Li S, Morita H, Altunbulakli C, et al. Distribution of ACE2, CD147, CD26, and other SARS-CoV-2 associated molecules in tissues and immune cells in health and in asthma, COPD, obesity, hypertension, and COVID-19 risk factors. Allergy. 2020; 7511: 2829–2845. https://doi.org/10.1111/all.14429
- Hasan A, Paray BA, Hussain A, Qadir FA, Attar F, Aziz FM, Sharifi M, Derakhshankhah H, Rasti B, Mehrabi M, et al. A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. J Biomol Struct Dyn. 2021; 398: 3025–3033. https://doi.org/10.1080/07391102.2020.1754293
- Langnau C, Rohlfing AK, Gekeler S, Gunter M, Poschel S, Petersen-Uribe A, Jaeger P, Avdiu A, Harm T, Kreisselmeier KP, et al. Platelet activation and plasma levels of furin are associated with prognosis of patients with coronary artery disease and COVID-19. Arterioscler Thromb Vasc Biol. 2021; 416: 2080–2096. https://doi.org/10.1161/ATVBAHA.120.315698
- Lood C, Arve S, Ledbetter J, Elkon KB. TLR7/8 activation in neutrophils impairs immune complex phagocytosis through shedding of FcgRIIA. J Exp Med. 2017. 2147: 2103–2119.https://doi.org/10.1084/jem.20161512
- Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, Kurt-Jones EA, Ravid K, Freedman JE. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. 2014. 1245: 791–802.https://doi.org/10.1182/blood-2013-11-536003
- Li XK, Lu QB, Chen WW, Xu W, Liu R, Zhang SF, Du J, Li H, Yao K, Zhai D, et al. Arginine deficiency is involved in thrombocytopenia and immunosuppression in severe fever with thrombocytopenia syndrome. Sci Transl Med. 2018 Sep 19;10(459):eaat4162. https://doi.org/10.1126/scitranslmed.aat4162.
- FitzGerald GA, Smith B, Pedersen AK, Brash AR. Increased prostacyclin biosynthesis in patients with severe atherosclerosis and platelet activation. N Engl J Med. 1984. 31017: 497–506.https://doi.org/10.1056/NEJM198404263101701
- Boilard E, Pare G, Rousseau M, Cloutier N, Dubuc I, Levesque T, Borgeat P, Flamand L. Influenza virus H1N1 activates platelets through FcgammaRIIA signaling and thrombin generation. Blood. 2014. 12318: 2854–2863.https://doi.org/10.1182/blood-2013-07-515536
- Nazy I, Jevtic SD, Moore JC, Huynh A, Smith JW, Kelton JG, Arnold DM. Platelet-activating immune complexes identified in critically ill COVID-19 patients suspected of heparin-induced thrombocytopenia. J Thromb Haemost. 2021. 195: 1342–1347.https://doi.org/10.1111/jth.15283
- WHOREAfC-TW G, JAC S, Murthy S, JV D, AS S, Villar J, DC A, Annane D, LCP A, Berwanger O, et al. Association between administration of systemic corticosteroids and mortality among critically Ill patients with COVID-19: a meta-analysis. JAMA. 2020; 324: 1330–1341. https://doi.org/10.1093/eurheartj/ehaa254
- Cano EJ, Fonseca Fuentes X, Corsini Campioli C, O’Horo JC, Abu Saleh O, Odeyemi Y, Yadav H, Temesgen Z. Impact of corticosteroids in coronavirus disease 2019 outcomes: systematic review and meta-analysis. Chest. 2021. 1593: 1019–1040.https://doi.org/10.1016/j.chest.2020.10.054
- Flisiak R, Jaroszewicz J, Rogalska M, Lapinski T, Berkan-Kawinska A, Bolewska B, Tudrujek-Zdunek M, Kozielewicz D, Rorat M, Leszczynski P, et al. Tocilizumab improves the prognosis of COVID-19 in patients with high IL-6. J Clin Med. 2021 Apr 9;10(8):1583. https://doi.org/10.390/jcm10081583
- Morris T, Stables M, Hobbs A, de Souza P, Colville-Nash P, Warner T, Newson J, Bellingan G, Gilroy DW. Effects of low-dose aspirin on acute inflammatory responses in humans. J Immunol. 2009. 1833: 2089–2096.https://doi.org/10.4049/jimmunol.0900477
- Müller CD, Karl N, Ziebuhr J, Pleschka S. D, L-lysine acetylsalicylate + glycine impairs coronavirus replication. J Antivir Antiretrovir. 2016; 084: https://doi.org/10.4172/jaa.1000151
- Glatthaar-Saalmuller B, Mair KH, Saalmuller A. Antiviral activity of aspirin against RNA viruses of the respiratory tract-an in vitro study. Influenza Other Respir Viruses. 2017; 11: 85–92
- Carestia A, Davis RP, Grosjean H, Lau MW, Jenne CN. Acetylsalicylic acid inhibits intravascular coagulation during Staphylococcus aureus-induced sepsis in mice. Blood. 2020. 13515: 1281–1286.https://doi.org/10.1182/blood.2019002783
- Falcone M, Russo A, Cangemi R, Farcomeni A, Calvieri C, Barilla F, Scarpellini MG, Bertazzoni G, Palange P, Taliani G, et al. Lower mortality rate in elderly patients with community-onset pneumonia on treatment with aspirin. J Am Heart Assoc. 2015; 41: e001595. https://doi.org/10.1161/JAHA.114.001595
- Ouyang Y, Wang Y, Liu B, Ma X, Ding R. Effects of antiplatelet therapy on the mortality rate of patients with sepsis: a meta-analysis. J Critical Care. 2019; 50: 162–168. https://doi.org/10.1016/j.jcrc.2018.12.004
- Panka BA, de Grooth HJ, Spoelstra-de Man AM, Looney MR, Tuinman PR. Prevention or treatment of ards with aspirin: a review of preclinical models and meta-analysis of clinical studies. Shock. 2017. 471: 13–21.https://doi.org/10.1097/SHK.0000000000000745
- Chow JH, Khanna AK, Kethireddy S, Yamane D, Levine A, Jackson AM, McCurdy MT, Tabatabai A, Kumar G, Park P. et al. Aspirin use is associated with decreased mechanical ventilation, intensive care unit admission, and in-hospital mortality in hospitalized patients with coronavirus disease 2019. Anesthesia and analgesia. 2021; 132: 930–941
- Liu Q, Huang N, Li A, Zhou Y, Liang L, Song X, Yang Z, Zhou X. Effect of low-dose aspirin on mortality and viral duration of the hospitalized adults with COVID-19. Medicine (Baltimore). 2021. 1006: e24544.https://doi.org/10.1097/MD.0000000000024544
- Viecca M, Radovanovic D, Forleo GB, Santus P. Enhanced platelet inhibition treatment improves hypoxemia in patients with severe Covid-19 and hypercoagulability. A case control, proof of concept study. Pharmacol Res. 2020; 158: 104950. https://doi.org/10.1016/j.phrs.2020.104950
- Marino M, Scuderi F, Ponte E, Maiuri MT, De Cristofaro R, Provenzano C, Rose-John S, Cittadini A, Bartoccioni E. Novel path to IL-6 trans-signaling through thrombin-induced soluble IL-6 receptor release by platelets. J Biol Regul Homeost Agents. 2013; 27: 841–852
- Dzeshka MS, Shantsila A, Lip GY. Effects of aspirin on endothelial function and hypertension. Curr Hypertens Rep. 2016. 1811: 83.https://doi.org/10.1007/s11906-016-0688-8
- Jung SB, Kim CS, Naqvi A, Yamamori T, Mattagajasingh I, Hoffman TA, Cole MP, Kumar A, Dericco JS, Jeon BH, et al. Histone deacetylase 3 antagonizes aspirin-stimulated endothelial nitric oxide production by reversing aspirin-induced lysine acetylation of endothelial nitric oxide synthase. Circ Res. 2010; 1077: 877–887. https://doi.org/10.1161/CIRCRESAHA.110.222968
- Merzon E, Green I, Vinker S, Golan-Cohen A, Gorohovski A, Avramovich E, Frenkel-Morgenstern M, Magen E. The use of aspirin for primary prevention of cardiovascular disease is associated with a lower likelihood of COVID-19 infection. FEBS J. 2021; https://doi.org/10.1111/febs.15784
- RECOVERY Collaborative Group, Horby PW, Pessoa-Amorim G, Staplin N, Emberson JR, Campbell M, Spata E, Peto L, Brunskill NJB, Tiberi S, et al. Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. medRxiv 2021.06.08.21258132; https://doi.org/10.1101/2021.06.08.21258132.
- Kunutsor SK, Laukkanen JA. Incidence of venous and arterial thromboembolic complications in COVID-19: a systematic review and meta-analysis. Thromb Res. 2020; 196: 27–30. https://doi.org/10.1016/j.thromres.2020.08.022
- Jimenez D, Garcia-Sanchez A, Rali P, Muriel A, Bikdeli B, Ruiz-Artacho P, Le Mao R, Rodriguez C, Hunt BJ, Monreal M. Incidence of VTE and bleeding among hospitalized patients with coronavirus disease 2019: a systematic review and meta-analysis. Chest. 2021. 1593: 1182–1196.https://doi.org/10.1016/j.chest.2020.11.005