
Abstract
Protease-activated receptor 4 (PAR4) is a promising drug target to improve the efficacy/safety window of antiplatelet agents. The native peptide GYPGQV, and the more-potent peptide AYPGKF, are PAR4-specific activators. However, these PAR4 agonist peptides (APs) elicit an agonist response, for example, platelet aggregation, at concentrations of 50 to 1000 µM in platelet-function assays, thereby limiting their utility to monitor the pharmacodynamic effects of PAR4 antagonists over a wide concentration range. Improved pharmacodynamic assays are needed for clinical development of PAR4 antagonists. We attempted to identify potent PAR4 APs to aid development of robust assays for optimization of PAR4 antagonists. Using an AYPG-based biased phage-display peptide library approach followed by chemical peptide optimization, A-Phe(4-F)-PGWLVKNG was identified. This peptide demonstrated an EC50 value of 3.4 µM in a platelet-aggregation assay, which is 16-fold more potent than AYPGKF. Using this new PAR4 AP, a platelet-rich plasma-aggregation assay using light-transmission aggregometry was developed and validated in a series of precision and reproducibility tests. PAR4 antagonist responses to PAR4 AP A-Phe(4-F)-PGWLVKNG (12.5 µM to 100 µM) were subsequently evaluated in this assay in vitro and ex vivo in a human study using BMS-986120, a PAR4 antagonist that entered clinical studies.
Introduction
In the past two decades, platelet thrombin receptors have become new targets for pharmaceutical research [Citation1,Citation2]. Human platelets express two distinct G-protein coupled thrombin receptors: protease-activated receptor-1 (PAR1) and protease-activated receptor-4 (PAR4) [Citation3,Citation4]. PAR1 and PAR4 mediate early and late-stage thrombin actions on human platelets, respectively, and thus are believed to differ in their roles in hemostasis and thrombosis [Citation2,Citation5,Citation6]. In 2014, an orally active PAR1 antagonist, vorapaxar, was approved for the reduction of thrombotic cardiovascular events in patients with a history of myocardial infarction or with peripheral arterial disease. However, similar to other current antiplatelet agents, vorapaxar treatment caused an increased risk of bleeding, thereby limiting its clinical utility [Citation7,Citation8,Citation9]. There remains a critical need to discover and develop new antiplatelet agents that can achieve efficacy with a reduced bleeding risk compared with currently marketed antiplatelet agents.
Recently, we discovered an orally active, potent, selective, and reversible PAR4 antagonist, BMS-986120 [Citation5]. In a nonhuman-primate model, BMS-986120 exhibited robust antithrombotic efficacy and markedly lower bleeding risk when compared head-to-head with clopidogrel [Citation5]. Our results suggest that PAR4 antagonists hold promise for addressing unmet medical need with a wider efficacy/safety window.
A variety of ex vivo platelet-function tests have been applied and reported previously to evaluate the pharmacodynamic effect of antiplatelet agents [Citation10]. Among these, light-transmission aggregometry (LTA) using platelet-rich plasma (PRP) is a standard method used to assess platelet function and drug effectiveness [Citation11]. To facilitate clinical development of PAR4 antagonists, we sought to develop and validate an ex vivo PRP LTA platelet-aggregation assay that is suitable for testing PAR4 activation and inhibition in clinical studies.
As thrombin activates both PAR1 and PAR4, interrogating receptor-specific function of PAR1 and PAR4 relies largely on synthetic agonist peptides (APs) that mimic receptor activation by thrombin. Based on the fact that PAR1 and PAR4 activation is initiated by thrombin cleavage of the N-terminus of the receptor that unmasks the tethered ligand sequence at the new amino-terminus [Citation3], synthetic peptides derived from the tethered ligand sequence of PAR1 (SFLLRN) [Citation12,Citation13] or PAR4 (GYPGQV) [Citation4,Citation14] were found to induce signaling events independent of receptor cleavage by thrombin. Through extensive library screening and structure–activity relationship studies, several potent and selective PAR1 APs (also called thrombin receptor activation peptides [TRAPs]) have been identified [Citation15–19]. These PAR1 APs contain 5–14 amino acid residues, mimicking the tethered ligand sequence of PAR1 [Citation20]. The most potent PAR1 AP achieved an EC50 value of 10 nM in a platelet-aggregation assay, which is approximately 1000-fold more potent than the original PAR1 AP [Citation19]. In contrast, the published PAR4 AP GYPGQV, or its rodent counterpart GYPGKF, is less potent and requires up to 1 mM concentration to induce a full platelet-aggregation response [Citation14,Citation21]. Subsequent peptide optimization led to the discovery of AYPGKF as a PAR4 AP with 10-fold improvement of potency compared with the original peptide [Citation22]. It has been shown that PAR1 AP or PAR4 AP, when used in vast molar excess over receptor concentration, can evoke many of the effects of the thrombin-mediated tethered ligand mechanisms in platelets [Citation16,Citation20,Citation23,Citation24]. Therefore, these receptor-specific APs have been widely used to characterize PAR1- and PAR4-specific signaling events and functions in vitro [Citation2,Citation6,Citation20].
As the most widely used PAR4 AP, AYPGKF exhibits only modest potency, requiring approximately 100 µM to induce full platelet aggregation. Its use in diagnostic tests may be limited by solubility, or potentially nonspecific activity, when much higher concentrations of PAR4 AP are desired to track a full range of pharmacodynamic effects of PAR4 antagonists over a wide dose range. Thus, a more-potent or more-selective PAR4 AP would be desirable to overcome the limitations of the classical PAR4 agonists.
Early in our PAR4 antagonist discovery program, we initiated efforts to discover a more-potent PAR4 AP using an AYPG-based biased phage-display peptide library approach. Subsequent optimization of selected phage clones using truncations as well as positional scanning with both natural and unnatural amino acids resulted in the discovery of A-Phe(4-F)-PGWLVKNG, which showed 16-fold improvement of potency compared with AYPGKF in platelet-aggregation assays. Using the PAR4 AP A-Phe(4-F)-PGWLVKNG, we developed in vitro assays to assist optimization of our small-molecule PAR4 antagonists, which led to the discovery of BMS-986120 [Citation5]. Subsequently, we developed and validated an LTA platelet-aggregation assay and determined PAR4 AP A-Phe(4-F)-PGWLVKNG concentrations required to assess the pharmacodynamic effects of BMS-986120 in clinical studies.
Materials and methods
Phage-display peptide library construction and panning, peptide synthesis, preparation of washed platelets, and PAR4 AP characterization using a calcium mobilization assay and 96-well platelet-aggregation assay are described in the Supplementary Materials.
Preparation of human PRP
Fresh blood was drawn by antecubital venipuncture from healthy volunteers who were free of exposure to antiplatelet drugs following institutionally informed consent. Samples were anticoagulated with sodium citrate. Following centrifugation of the blood at 250 xg for 10 minutes at room temperature, PRP was collected, and the remaining sample was centrifuged at 2000 xg for 10 minutes at room temperature to prepare platelet-poor plasma.
LTA platelet-aggregation assay
Platelet aggregation was tested using either a Chrono-Log Model 700 Whole Blood/Optical Lumi-Aggregometer (Chrono-Log Corporation, Havertown, PA, USA) or a Bio/Data Model PAP-8E (Bio/Data Corp., Horsham, PA, USA). PAR4 AP A-Phe(4-F)-PGWLVKNG or AYPGKF (GenScript, Piscataway, NJ, USA) were prepared as 20X stock solutions using Hanks’ Balanced Saline. Platelet aggregation was initiated by addition of 25 µL of PAR4 AP A-Phe(4-F)-PGWLVKNG to 475 µL of PRP and monitored for 10 minutes, according to the manufacturer’s instructions.
To test the inhibitory effect of PAR4 antagonist in vitro, BMS-9861205 was prepared as 200X stock solutions in dimethyl sulfoxide, diluted to 20X working solution in platelet-poor plasma, and then the working solution was added to PRP at a 1:20 ratio and incubated at 37°C for 60 minutes before aggregation was initiated by addition of PAR4 AP A-Phe(4-F)-PGWLVKNG.
BMS-986120 concentration responses ex vivo were tested in a phase 1 clinical trial described in detail by Ismat et al [Citation25]. Briefly, BMS-986120 was administered orally to healthy participants at 0.3, 1, 3, 10, 30, 75, and 180 mg doses (n = 6/dose), and blood samples were collected at 1, 2, 3, 4, 12, 24, 48, 72, 96, and 144 hours after dosing. PRP was prepared, stimulated with PAR4 AP A-Phe(4-F)-PGWLVKNG at 12.5, 25, 50, and 100 µM, and monitored using LTA.
Results
Phage-display library design and selection for PAR4-binding peptides
A phage-display approach was used to identify novel PAR4 APs. For this purpose, a biased peptide library consisting of AYPGXXXXXX, where X is a random natural amino acid, was constructed. The short peptide, AYPG (peptide 16; ), was derived from the known PAR4 AP sequence AYPGKF (peptide 1; ) [Citation22] and was found to retain modest activity for inducing calcium mobilization in HEK293-PAR4 cells. Six random residues were chosen to maximize library diversity while limiting sequence length to 10 residues. The library quality was verified by DNA sequencing of 78 randomly picked clones, and 20% of the clones had inserts with apparently random distribution of residues in the library segment. The total number of library clones was approximately 109, thus yielding an effective library size of approximately 2 × 108.
Table I. Summary of PAR4 binding peptides and their agonist activities in FLIPR calcium mobilization assay and 96-well platelet aggregation assay.
To focus the selection on PAR4 agonist activity, a series of unique panning steps were employed. As PAR4 protein was unavailable in purified form, cell membranes from PAR4-expressing HEK293-PAR4 cells [Citation5] were used to pan against the biased library. Pre-adsorption was carried out with the parental HEK293 cell membranes and with a purified integrin to minimize nonspecific binding. PAR4-specific binding was eluted competitively with AYPGKF peptide. A second pan was carried out in which total phage binding was eluted nonspecifically with acid or recovered by direct E. coli infection. As controls, a pair of pans were performed using parental HEK293 membranes and elution with either AYPGKF peptide or nonspecifically with direct E. coli infection without peptide elution. After three cycles of panning, DNA sequencing was used to identify clones with peptide inserts. Table SI summarizes the number of clones with inserts identified from each of the four pans.
To find new PAR4 APs, results from the four pans were compared, and unique peptides from HEK293-PAR4 panning and AYPGKF elution were identified. Out of 28 potential clones obtained from HEK293-PAR4 panning and AYPGKF elution, 6 were unique and specific for PAR4 binding based on the panning properties (Table SII).
PAR4 AP lead optimization
The six candidate peptides (peptide 2 to peptide 7; ) were synthesized and tested for PAR4 agonist activity in a calcium-mobilization assay. AYPGWLVKNG (peptide 7; EC50, 4.8 µM; ) was significantly more potent than the others, including AYPGKF (peptide 1; EC50, 61 µM; ). Subsequent structure–activity relationship studies of peptide 7 by replacing the Tyr2 phenolic hydroxyl with its “classical bioisostere” Phe(4-F) fluorine [Citation26] had a modest improvement in potency (peptide 8; EC50, 2.3 µM; ). Overall, peptide 8 (A-Phe(4-F)-PGWLVKNG) showed a 27-fold potency improvement in the calcium mobilization assay and a 15-fold improvement in a 96-well platelet-aggregation assay compared with AYPGKF.
Additional truncation and positional scans using natural and unnatural amino acids were performed to further investigate the structure–activity relationships of peptide 8. Incremental residue truncations at the C-terminus of the lead peptide, A-Phe(4-F)-PGWLVKNG, indicated that the minimal sequence required to maintain optimal potency is the 7-mer, A-Phe(4-F)-PGWLV (peptide 11; ). This 7-mer and the unmodified peptide AYPGWLV (peptide 12; ) were used for further analysis. Notably, a 15-fold loss of activity upon truncation of Trp5 (peptide 15 and 16; ) underscores the importance of this residue. Furthermore, replacement of the Leu6 and Val7 residues with Ala in peptide 12 caused a similar degree of loss of activity to that seen upon truncation of those 2 residues in peptide 11 (peptide 17 and 18 versus peptide 13 and 14; ), suggesting that the alkyl side chains of those residues may provide additional contributions to receptor activation.
The importance of the L-configuration at each amino acid position of peptide 11 was tested by inverting the configuration from L to D at each position. In all cases, the D-configuration abolished activity (data not shown), clearly demonstrating the requirement for the natural L-configuration for agonist activity.
Positional scans using natural and unnatural amino acids were carried out within the key aromatic positions Tyr2 or Phe(4-F)2 (Table SIII), the conformationally interesting Pro3-Gly4 dipeptide (Table SIV and SV), Trp5 (Table SVI), and the C-terminal extended positions Leu6 and Val7 (Table SVII and SVIII). Several aromatic substitutions were well accepted at the Tyr2 position, in addition to the previously mentioned 4-fluorophenylalanine (Table SIII). Single and multiple halogen phenyl ring substitutions resulted in modest potency enhancements, as did several other replacements at the ortho and meta positions, suggesting that the para position is not the sole contributor to activity improvements (Table SIII). The Trp5 position tolerated a variety of aromatic replacements, with the bioisosteric benzothiophene (Bzt), the penta-fluorophenylalanine [Phe(penta-F)], and the 1- and 2-naphthylalanine replacements fully retaining or slightly improving activity (Table SVI). In contrast to the tolerance on the substitution of positions 2 and 5, the Pro3 position only accepted replacement of the 5-member pyrrolidine ring with the smaller and larger ring homologues azetidine (Azt) and piperidine (Pip), respectively (Table SIV). Notably, the conformational importance of a tertiary amide bond at this position seemed to be supported by the retention of activity seen with N-Methyl-Gly and N-Methyl-Ala analogues. The Gly4 position tolerated no side chain except for the methyl side chain of Ala (Table SV), suggesting that Gly4 may be an important conformational switch position requiring greater flexibility.
Through structure–activity relationship studies, A-Phe(4-F)-PGWLVKNG (peptide 8) emerged as the lead peptide. Furthermore, the peptide’s sequence composition and physicochemical properties, especially its improved solubility, rendered it amenable to synthesis scale-up, reinforcing its selection as the lead peptide.
Characterization of the lead PAR4 AP composed of A-Phe(4-F)-PGWLVKNG
The lead peptide A-Phe(4-F)-PGWLVKNG was further characterized in comparison to the reference peptide AYPGKF (peptide 1; ). In a calcium mobilization assay using HEK293-PAR4 cells, A-Phe(4-F)-PGWLVKNG elicited calcium mobilization profiles similar to those induced by AYPGKF (). In contrast, when tested at 11 µM (at 5X its PAR4 EC50 value) in parental HEK293 cells that only express PAR1 [Citation27, A-Phe(4-F)-PGWLVKNG had no activity ().
Figure 1. A-Phe(4-F)-PGWLVKNG peptide induced PAR4 activation in HEK293-PAR4 cells and human platelets. (a) Representative FLIPR tracing of A-Phe(4-F)-PGWLVKNG- and AYPGKF-induced calcium mobilization in HEK293-PAR4 cells. To control for the cell population in each experiment and between conditions, both peptides were used at EC80. (b) Representative FLIPR tracing of A-Phe(4-F)-PGWLVKNG (11 µM) -induced calcium mobilization in HEK293-PAR4 cells versus HEK293 parental cells. (c) Representative FLIPR tracing of A-Phe(4-F)-PGWLVKNG versus PAR1 AP (SFFLRR)-induced calcium mobilization in human platelets. Both peptides were used at 12.5 µM. Results from A, B, C represent data from n ≥ 2. (d) Comparison of a set of 178 small-molecule PAR4 antagonist activities in the calcium mobilization assay performed using HEK293-PAR4 cells stimulated with either A-Phe(4-F)-PGWLVKNG or AYPGKF at EC50. Correlation analysis was performed using GraphPad Prism software and the Spearman rs and P values are given.
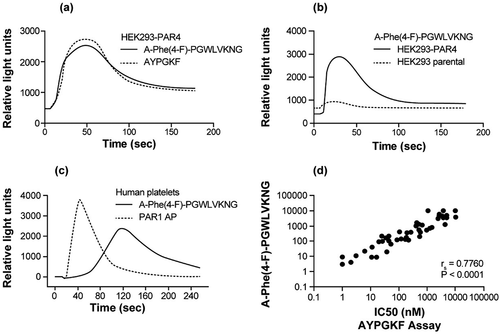
One of the hallmarks of PAR4 activation in human platelets is a slow and sustained calcium mobilization profile, while PAR1 activation elicits a fast and short-lived calcium signaling [Citation28,Citation29]. A-Phe(4-F)-PGWLVKNG, like other PAR4 APs including AYPGKF [Citation5], induced a calcium mobilization profile that is characteristic of PAR4 activation in human platelets, and is distinct from that induced by PAR1 AP (SFFLRR; ). This is consistent with previously published results showing that calcium mobilization profiles are similar when human platelets were activated by A-Phe(4-F)-PGWLVKNG or thrombin treated with a PAR1 inhibitor [Citation5].
A-Phe(4-F)-PGWLVKNG and AYPGKF were used to compare pharmacological responses of 178 small-molecule PAR4 antagonists in the calcium mobilization assay. These small-molecule PAR4 antagonists were derived from hits discovered via high-throughput screening of our compound collection for their ability to inhibit AYPGKF-induced calcium mobilization in the HEK293-PAR4 cell-based FLIPR assay. The results demonstrated a good correlation between the IC50 values derived from cells stimulated by A-Phe(4-F)-PGWLVKNG and AYPGKF (Spearman’s rank correlation coefficient rs = 0.7760; P < .0001; ).
Comparison of A-Phe(4-F)-PGWLVKNG and AYPGKF responses in LTA platelet-aggregation assay
We sought to develop a standard LTA platelet-aggregation assay using PRP and PAR4 AP A-Phe(4-F)-PGWLVKNG to induce platelet activation. Using the Chrono-Log LTA instrument, PAR4 AP A-Phe(4-F)-PGWLVKNG was tested side-by-side with AYPGKF. The two peptides induced concentration-dependent full aggregation responses but exhibited different potencies (). A-Phe(4-F)-PGWLVKNG induced platelet aggregation at an EC50 of 3.4 µM, which is 16-fold more potent than AYPGKF (EC50, 56 µM). The minimum concentration to reach maximum threshold response was 6.25 µM with A-Phe(4-F)-PGWLVKNG; in comparison, 100 µM of AYPGKF was the minimum concentration required to attain maximum threshold response. Based on these results, A-Phe(4-F)-PGWLVKNG was the preferred agonist to further develop PAR4 activation and inhibition assays for clinical use.
Figure 2. PAR4 AP evaluation in LTA platelet-aggregation assays. (a) PAR4 AP concentration responses in LTA platelet-aggregation assay. A-Phe(4-F)-PGWLVKNG or AYPGKF were tested side-by-side using the Chrono-Log instrument. The EC50 values were calculated from 4 parameter non-linear regression curve fit of 9 to 10 independent replicates using GraphPad Prism 7.02 software. (b) Comparison of Chrono-Log versus Bio/Data instruments in measuring human PRP responses to A-Phe(4-F)-PGWLVKNG. Results represent average of data from 5 donors. Error bars indicate standard error of the mean.
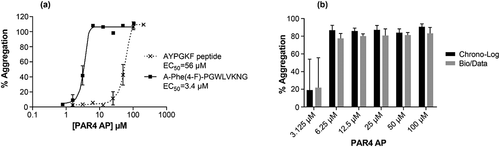
The performance of two commonly used LTA instruments, Chrono-Log Model 700 and Bio/Data PAP-8E, were compared using PRP samples from 5 donors and evaluated by CirQuest Labs (Memphis, TN, USA). The magnitude of aggregation responses to a concentration range of A-Phe(4-F)-PGWLVKNG were comparable using the two instruments and both showed a threshold of maximum response at 6.25 µM (). Together, these results suggest that A-Phe(4-F)-PGWLVKNG is an optimized PAR4 AP suitable for PAR4 platelet-function tests using common LTA instruments.
Validation of PAR4 platelet-function test using the LTA platelet-aggregation assay
To support the clinical utilization of the PAR4 platelet-function test using the LTA platelet-aggregation assay, a series of validation tests were performed at Pharmaceutical Product Development, LLC (Austin, TX, USA), a global contract research organization. PRP samples from four healthy participants prepared on four different days from freshly drawn blood were tested using different Chrono-Log aggregometer instruments (four in total) each day. The PAR4 AP, A-Phe(4-F)-PGWLVKNG, at 12.5 µM concentration, was used to induce a full platelet aggregation.
To evaluate instrument precision, PRP samples were run only on aggregometer 3 on day 1, aggregometers 1 and 3 on day 2, aggregometers 2 and 3 on day 3, and aggregometers 3 and 4 on day 4. Intra-assay precision was calculated based on the magnitude of aggregation from replicate runs using the same instrument on the same day for each of the donors, while inter-assay precision was calculated based on replicate runs on different days on the same instruments for each of the donors. Both intra-assay precision and inter-assay precision showed coefficient of variation values ranging from 3.2% to 5.4% and 3.2% to 5.8%, respectively ().
Table II. Summary of Chrono-Log LTA platelet-aggregation assay performance using PAR4 AP (A-Phe[4-F]-PGWLVKNG) tested at 12.5 μM.
To evaluate variation introduced by different laboratory technicians, analyst imprecision was tested by eight technicians using PRP samples from four donors collected on the same day. Each of the eight technicians tested samples from all four donors. The results of the magnitude of aggregation derived from each donor demonstrated a high level of consistency across all analysts, with a coefficient of variation value of 2.1% to 3.9% ().
Assay window precision and PRP sample stability were tested using PRP samples from four donors analyzed approximately 1, 2, and 3 h after blood collection. The results of the mean magnitude of aggregation using PAR4 AP A-Phe(4-F)-PGWLVKNG (12.5 μM) were comparable within the 3-h test window and showed a coefficient of variation value of 1.4% to 2.8% ().
The reference ranges of the mean magnitude of aggregation were tested using PRP samples from 20 donors and the values were assigned as plus and minus 2 standard deviations. The resulting mean percent aggregation induced by PAR4 AP A-Phe(4-F)-PGWLVKNG (12.5 μM) was within a range of 83% to 102% ().
The stability of working solutions of PAR4 AP A-Phe(4-F)-PGWLVKNG at 200, 500, 1000, and 2000 μM stored at −20°C and −80°C were evaluated at different time points by CirQuest Lab (Memphis, TN, USA) using the Chrono-Log LTA platelet-aggregation assay. The coefficient of variation of maximal aggregation for all working solutions stored for up to 6 months was within 0% to 13% ().
Table III. Summary of working solution stability of PAR4 AP for platelet aggregation.
PAR4 antagonist responses in LTA platelet-aggregation assay
In vitro inhibitory activity of a potent and selective PAR4 antagonist, BMS-986120, was evaluated using the Chrono-Log LTA instrument and PAR4 AP A-Phe(4-F)-PGWLVKNG. At 12.5 µM PAR4 AP A-Phe(4-F)-PGWLVKNG, which induced full platelet aggregation in samples of all 6 donors, BMS-986120 completely inhibited platelet activation (). For each individual donor, the inhibitory effect of BMS-986120 appeared to be dichotomous, with a steep concentration response curve (). The concentration response of BMS-986120 was right-shifted when 50 µM of PAR4 AP A-Phe(4-F)-PGWLVKNG was used ().
Figure 3. Concentration-dependent inhibition of PAR4 AP (A-Phe(4-F)-PGWLVKNG) -induced platelet aggregation by BMS-986120 using LTA platelet-aggregation assay. (a) In vitro concentration responses of BMS-986120 to 12.5 µM PAR4 AP A-Phe(4-F)-PGWLVKNG from individual donors are shown. Isolated PRP was treated with BMS-986120 for 60 minutes and stimulated with 12.5 µM PAR4 AP A-Phe(4-F)-PGWLVKNG. (b) The average in vitro concentration responses and standard error of means of BMS-986120 are shown (n = 6 donors per group). Isolated PRP was treated with BMS-986120 for 60 minutes and stimulated with 12.5 µM and 50 µM PAR4 AP A-Phe(4-F)-PGWLVKNG. (c) Ex vivo concentration responses of BMS-986120 to PAR4 AP A-Phe(4-F)-PGWLVKNG are shown (n = 42 donors). BMS-986120 was administered to healthy volunteers as described in the method and PRP samples were collected and stimulated with PAR4 AP A-Phe(4-F)-PGWLVKNG at 12.5 µM, 25 µM, 50 µM and 100 µM. (d) The calculated IC50 values from in vitro (B) and ex vivo (C) tests are displayed. The IC50 values are indicated and were derived from four parameter nonlinear regression curve fit using GraphPad Prism 7.02 software. The dashed line indicates historical in vitro potency of BMS-986120 in PRP-aggregation assay stimulated with γ-thrombin (γ-Thr).5
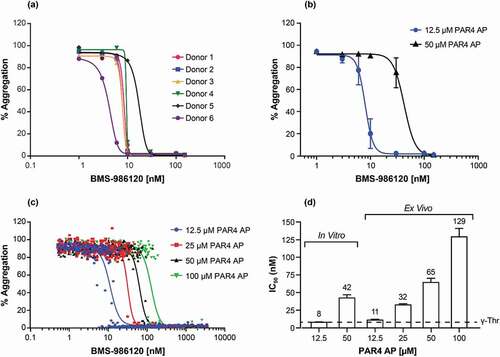
A broader range of the PAR4 AP A-Phe(4-F)-PGWLVKNG at 12.5, 25, 50, and 100 µM was then selected to measure the pharmacodynamic effect of BMS-986120 in a phase 1 study. BMS-986120 was administered orally to normal healthy volunteers at 0.3, 1, 3, 19, 30, 75, and 180 mg doses and blood samples were processed to measure PRP aggregation responses ex vivo using the Chrono-Log LTA instrument. Platelet aggregation induced by the full range of PAR4 AP A-Phe(4-F)-PGWLVKNG concentrations were completely inhibited by BMS-986120 in a concentration-dependent manner (). The IC50 values of BMS-986120 shifted from 11 nM to 129 nM when PAR4 AP A-Phe(4-F)-PGWLVKNG concentrations increased from 12.5 µM to 100 µM (). It is noted that the IC50 values of BMS-986120 from the in vitro and ex vivo tests using 12.5 µM PAR4 AP A-Phe(4-F)-PGWLVKNG (8 nM and 11 nM, ) closely match that previously obtained in a PRP-aggregation assay using γ-thrombin (7.3 nM) [Citation5], a thrombin fragment that specifically cleaves PAR4 but not PAR1 [Citation30].
Discussion
As described herein, the new PAR4 AP, A-Phe(4-F)-PGWLVKNG, has the characteristics required to be an important tool for developing pharmacodynamic assays to monitor the activity of PAR4 antagonists. To overcome the limitation of the modest potency of previously described PAR4 APs, including AYPGKF, we sought to discover new PAR4 APs using a phage-display approach, which was based on a biased library comprised of a weakly active N-terminal tetrapeptide AYPG and 6 randomized residues extended at the C-terminus. A full combinatorial library was cloned, expressed, and panned against membranes derived from HEK293-PAR4 cells. Pre-adsorption with the parental HEK293 membranes was used to remove non-PAR4 binders. Moreover, this pre-adsorption step using HEK293 membranes, which endogenously express PAR1 [Citation27], eliminated PAR1 binders, as was demonstrated by PAR4 selectivity from the phage-display hits and subsequently optimized peptides.
A lead peptide, AYPGWLVKNG, identified from the phage-display approach, displayed a more than 10-fold improvement of potency over the reference PAR4 AP, AYPGKF. Subsequent structure–activity relationship optimization led to the discovery of A-Phe(4-F)-PGWLVKNG. In a standard LTA platelet-aggregation assay, A-Phe(4-F)-PGWLVKNG achieved a threshold of maximum response at 6.25 µM and an IC50 value of 3.4 µM, which is a 16-fold improvement over AYPGKF. Supporting its selectivity, A-Phe(4-F)-PGWLVKNG stimulated calcium mobilization in HEK293-PAR4 cells but not in the parental cells that endogenously express PAR1 but not PAR4. It is noteworthy that replacement of Tyr2 for Phe(4-F) in A-Phe(4-F)-PGWLVKNG did not result in a loss of PAR4 selectivity. In contrast, this substitution affected PAR4 agonist selectivity in GYPGKF, an analogue precursor of AYPGKF [Citation22].
Importantly, A-Phe(4-F)-PGWLVKNG elicited a slow and sustained calcium-signaling profile in human platelets, a hallmark of PAR4 activation, similar to that of the natural activator, thrombin. Furthermore, the pharmacological activities of 178 small-molecule PAR4 antagonists were well correlated in calcium-mobilization assays when cells were stimulated with either A-Phe(4-F)-PGWLVKNG or AYPGKF. These results indicate that A-Phe(4-F)-PGWLVKNG is an optimized and selective PAR4 AP that can be used to develop robust PAR4-specific assays, such as LTA platelet aggregation, P-selectin expression, or other point-of-care tests [Citation10,Citation31]. A-Phe(4-F)-PGWLVKNG was used to develop a standardized LTA platelet-aggregation assay. In this assay, samples tested within 3 h of blood collection demonstrated intra-assay and inter-assay precision and inter-analyst precision with coefficients of variation less than 6% across all parameters.
This standardized LTA platelet-aggregation assay was subsequently used to test the pharmacodynamic effect of BMS-986120. To better understand the inhibitory potential for BMS-986120, two concentrations of PAR4 AP A-Phe(4-F)-PGWLVKNG (12.5 and 50 µM) were used to stimulate platelet aggregation. Despite 12.5 and 50 µM PAR4 AP A-Phe(4-F)-PGWLVKNG exceeding the threshold concentration (6.25 µM) required for maximal platelet aggregation, BMS-986120 completely inhibited platelet aggregation at both concentrations. However, the inhibition response of BMS-986120 was right-shifted with 50 µM PAR4 AP A-Phe(4-F)-PGWLVKNG. This result is consistent with our previous observation in ex vivo monkey studies, suggesting that BMS-986120 antagonism is surmountable [Citation5].
A significant advantage of PAR4 AP A-Phe(4-F)-PGWLVKNG is that drug activity can be tracked over the full concentration range in early clinical studies of agents such as BMS-986120. The ability to use higher concentrations of PAR4 AP A-Phe(4-F)-PGWLVKNG (up to 100 µM) in a human platelet-aggregation assay following single ascending-dose administration of 0.3 to 180 mg BMS-986120 [Citation25] indicates the ability to effectively monitor platelet activity and any other drug-related effects even at the highest drug concentration. More specifically, use of higher concentrations of PAR4 AP A-Phe(4-F)-PGWLVKNG keeps the platelet-aggregation assay in dynamic range when drug concentration is too high. These studies would not be feasible with AYPGKF, which has a relatively higher EC50 of 56 µM and would require up to 1.6 mM concentration in this assay to track the drug over the full concentration range. Furthermore, it is noted that the potency values of BMS-986120 measured in vitro were consistent with those values observed ex vivo in the clinical setting [Citation32].
To understand the connection of the effect of BMS-986120 in the PAR4 AP A-Phe(4-F)-PGWLVKNG assay to that in a thrombin assay, the IC50 values derived from the PAR4 AP A-Phe(4-F)-PGWLVKNG LTA platelet-aggregation assay were compared to those obtained by using γ-thrombin. The proteolytic product of native thrombin, γ-thrombin, lacks the hirudin-binding domain and selectively cleaves PAR4 without activating PAR1 or the coagulation process via the natural tethered ligand [Citation5,Citation29]. The IC50 value of BMS-986120 using γ-thrombin (7.3 nM) is similar to that obtained in vitro (8 nM) or ex vivo (11 nM) using 12.5 µM of PAR4 AP A-Phe(4-F)-PGWLVKNG. Therefore, platelet activation induced by 12.5 µM PAR4 AP A-Phe(4-F)-PGWLVKNG most closely reflects PAR4 endogenous activity induced by thrombin cleavage.
The ex vivo response of BMS-986120 to 12.5 µM PAR4 AP A-Phe(4-F)-PGWLVKNG also tracked well to the antithrombotic efficacy of BMS-986120 in our previously reported monkey model of arterial thrombosis [Citation5]. Using PAR4 AP A-Phe(4-F)-PGWLVKNG at 12.5 µM, BMS-986120 at 0.2 mg/kg or 1 mg/kg caused partial or complete inhibition, respectively, of ex vivo whole-blood aggregation.5 These 0.2 mg/kg or 1 mg/kg doses caused dose-dependent, ex vivo, platelet responses of BMS-986120 corresponding to a partial or near-complete reduction of thrombus weights, respectively. Therefore, we hypothesize that the LTA platelet-aggregation assay using 12.5 µM PAR4 AP A-Phe(4-F)-PGWLVKNG might be a useful biomarker to predict the antithrombotic efficacy of BMS-986120 in humans. Further clinical studies are required to establish the predictive value of these PAR4 functional assays and define the utility of these assays to potentially guide treatment strategies.
Overall, our data support the use of this new PAR4 AP A-Phe(4-F)-PGWLVKNG in LTA platelet-aggregation assays to evaluate the pharmacodynamic effect of PAR4 antagonists in clinical studies.
Author contributions
C. Mapelli, D.A. Seiffert, C.S. Sum, R.M. Lawrence, Z. Wang, Y. Ni, and J. Yang designed the research. J. Hua and C.S. Sum performed the experiments. All authors analyzed the results of the experiments and reviewed and approved the manuscript. J. Yang, C. Mapelli, C.S. Sum, J. Hua, Z. Wang, and D.A. Seiffert wrote the manuscript.
Supplemental Material
Download PDF (370.2 KB)Acknowledgements
We thank Anne Marinier, Scott Priestley, David Gordon, Ruth Wexler, Victor Guarino, and the entire PAR4 team for the discovery of BMS-986120; Michael G. Kornacker, Douglas J. Riexinger, Karen S. Hartl, and Nick J Allegretto for peptide synthesis and characterization; Thomas Connolly, Saurabh Gupta, Mary Ellen Cvijic, and Liang Schweizer for their input on the platelet-function assay and FLIPR assay; Pharmaceutical Product Development, LLC (Austin, TX, USA) for conducting assay performance and clinical samples tests; CirQuest Labs (Memphis, TN, USA) for performing comparison studies using Chrono-Log and Bio/Data instruments and working solution stability studies. Professional medical writing and editorial assistance was provided by Scott Forbes, PhD, of Oxford PharmaGenesis Inc. (Newtown, PA, USA), and was funded by Bristol Myers Squibb.
Disclosure statement
Z. Wang, J. Hua, R.M. Lawrence, and D.A. Seiffert are employees of Bristol Myers Squibb; J. Yang, C. Mapelli, C.S. Sum, and Y. Ni are former employees of Bristol Myers Squibb.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Franchi F, Rollini F, Park Y, Angiolillo DJ. Novel antiplatelet agents: the current state and what is coming down the pike. Prog Cardiovasc Dis 2015;583:267–277. DOI:10.1016/j.pcad.2015.08.009
- Yang J, Xu K, Seiffert D. Challenges and promises of developing thrombin receptor antagonists. Recent Pat Cardiovasc Drug Discov 2010;53:162–170. https://www.ingentaconnect.com/content/ben/prc/2010/00000005/00000003/art00002
- Coughlin SR. Protease-activated receptors and platelet function. Thromb Haemost 1999;822:353–356. DOI:10.1055/s-0037-1615853
- Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest 1999;1036:879–887. DOI:10.1172/JCI6042
- Wong PC, Seiffert D, Bird JE, Watson CA, Bostwick JS, Giancarli M, Allegretto N, Hua J, Harden D, Guay J, et al. Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci Transl Med 2017;9371. DOI:10.1126/scitranslmed.aaf5294
- French SL, Hamilton JR. Protease-activated receptor 4: from structure to function and back again. Br J Pharmacol 2016;17320:2952–2965. DOI:10.1111/bph.13455
- Eikelboom JW, Hirsh J, Spencer FA, Baglin TP, Weitz JI. Antiplatelet drugs - Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2012;1412:e89S–e119S. DOI:10.1378/chest.11-2293
- Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, Fox KAA, Lipka LJ, Liu X, Nicolau JC, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med 2012;36615:1404–1413. DOI:10.1056/NEJMoa1200933
- Tricoci P, Huang Z, Held C, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med 2012;3661:20–33. DOI:10.1056/NEJMoa1109719
- Paniccia R, Priora R, Liotta AA, Abbate R. Platelet function tests: a comparative review. Vasc Health Risk Manag 2015;11:133–148. DOI:10.2147/VHRM.S44469
- Born GV. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 1962;1944832:927–929. DOI:10.1038/194927b0
- Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991;646:1057–1068. DOI:10.1016/0092-8674(91)90261-V
- Vandendries ER, Hamilton JR, Coughlin SR, Furie B, Furie BC. Par 4 is required for platelet thrombus propagation but not fibrin generation in a mouse model of thrombosis. Proc Natl Acad Sci U S A 2007;1041:288–292. DOI:10.1073/pnas.0610188104
- Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW, Foster DC, et al. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A 1998;9512:6642–6646. DOI:10.1073/pnas.95.12.6642
- Chen J, Bernstein HS, Chen M, Wang L, Ishii M, Turck CW, Coughlin SR, et al. Tethered ligand library for discovery of peptide agonists. J Biol Chem 1995;40:23398–23401. DOI:10.1074/jbc.270.40.23398
- Seiler SM, Peluso M, Tuttle JG, Pryor K, Klimas C, Matsueda GR, Bernatowicz MS, et al. Thrombin receptor activation by thrombin and receptor-derived peptides in platelet and CHRF-288 cell membranes: receptor-stimulated GTPase and evaluation of agonists and partial agonists. Mol Pharmacol 1996;491:190–197.
- Natarajan S, Riexinger D, Peluso M, Seiler SM. ‘Tethered ligand’ derived pentapeptide agonists of thrombin receptor: a study of side chain requirements for human platelet activation and GTPase stimulation. Int J Pept Protein Res 1995;452:145–151. DOI:10.1111/j.1399-3011.1995.tb01033.x
- Scarborough RM, Naughton MA, Teng W, Hung DT, Rose J, Vu TK, Wheaton VI, Turck CW, Coughlin SR, et al. Tethered ligand agonist peptides. Structural requirements for thrombin receptor activation reveal mechanism of proteolytic unmasking of agonist function. J Biol Chem 1992;26719:13146–13149. DOI:10.1016/S0021-9258(18)42184-9
- Feng DM, Veber DF, Connolly TM, Condra C, Tang MJ, Nutt RF. Development of a potent thrombin receptor ligand. J Med Chem 1995;3820:4125–4130. DOI:10.1021/jm00020a029
- Hollenberg MD. Proteinase-activated receptors: tethered ligands and receptor-activating peptides. Drug Dev Res 2003;594:336–343. DOI:10.1002/ddr.10301
- Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV, Tam C, Coughlin SR, et al. A dual thrombin receptor system for platelet activation. Nature 1998;3946694:690–694. DOI:10.1038/29325
- Faruqi TR, Weiss EJ, Shapiro MJ, Huang W, Coughlin SR. Structure-function analysis of protease-activated receptor 4 tethered ligand peptides. Determinants of specificity and utility in assays of receptor function. J Biol Chem 2000;27526:19728–19734. DOI:10.1074/jbc.M909960199
- Vassallo RR Jr., Kieber-Emmons T, Cichowski K, Brass LF. Structure-function relationships in the activation of platelet thrombin receptors by receptor-derived peptides. J Biol Chem 1992;2679:6081–6085. DOI:10.1016/S0021-9258(18)42664-6
- Huang RS, Sorisky A, Church WR, Simons ER, Rittenhouse SE. “Thrombin” receptor-directed ligand accounts for activation by thrombin of platelet phospholipase C and accumulation of 3-phosphorylated phosphoinositides. J Biol Chem 1991;26628:18435–18438. DOI:10.1016/S0021-9258(18)55079-1
- Ismat FA, Ma X, Wang Z, Frost CE, Ni YG, Yang J. Abstract TMP91: phase I assessment of the safety, tolerability, pharmacokinetics and pharmacodynamics of the oral protease-activated receptor-4 antagonist BMS-986120. Stroke 2016;47Suppl 1: ATMP91. DOI:10.1161/str.47.suppl_1.tmp91
- Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem 2011;548:2529–2591. DOI:10.1021/jm1013693
- Kawabata A, Saifeddine M, Al-ani B, Hollenberg MD. Protease-activated receptors: development of agonists selective for receptors triggered by either thrombin (PAR1) or trypsin (PAR2). Proc West Pharmacol Soc 1997;40:49–51.
- Ishii K, Hein L, Kobilka B, Coughlin SR. Kinetics of thrombin receptor cleavage on intact cells. Relation to signaling. J Biol Chem 1993;26813:9780–9786. DOI:10.1016/S0021-9258(18)98415-2
- Covic L, Gresser AL, Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 2000;3918:5458–5467. DOI:10.1021/bi9927078
- Soslau G, Goldenberg SJ, Class R, Jameson B. Differential activation and inhibition of human platelet thrombin receptors by structurally distinct alpha-, beta- and gamma-thrombin. Platelets 2004;153:155–166. DOI:10.1080/0953710042000199848
- Sobieraj-Teague M, Eikelboom JW. Point-of-care testing for assessment of adequacy of oral antiplatelet therapy in patients with cardiovascular disease. Future Cardiol 2010;63:289–299. DOI:10.2217/fca.10.20
- Merali S, Wang Z, Frost C, Callejo M, Hedrick M, Hui L, Meadows Shropshire S, Xu K, Bouvier M, DeSouza MM, Yang J. New oral protease-activated receptor 4 antagonist BMS-986120: tolerability, pharmacokinetics, pharmacodynamics, and gene variant effects in humans. Platelets. 2022; Jun 26:1–0.