
Abstract
BMS-986120 is a novel first-in-class oral protease-activated receptor 4 (PAR4) antagonist exhibiting robust antithrombotic activity that has shown low bleeding risk in monkeys. We sought to assess pharmacokinetics, pharmacodynamics, and tolerability of BMS-986120 in healthy participants and platelet responses to BMS-986120 in participants carrying PAR4 A120T variants. Phase I, randomized, double-blind, placebo-controlled single-ascending-dose (SAD; N = 56) and multiple-ascending-dose (MAD; N = 32) studies were conducted. Exposure was approximately dose-proportional: maximum concentrations 27.3 and 1536 ng/mL, areas under the curve (AUC) to infinity of 164 and 15,603 h*ng/mL, and half-lives of 44.7 and 84.1 hours for 3.0 and 180 mg, respectively. The accumulation index suggested an ~2-fold AUC increase at steady state. Single doses of 75 and 180 mg BMS-986120 produced ≥80% inhibition of 12.5 μM PAR4 agonist peptide (AP)-induced platelet aggregation through at least 24 hours postdose, and doses ≥10 mg for ~7 days inhibited aggregation completely through 24 hours. No differences in PAR4-mediated platelet response were seen between AA120 versus TT120 PAR4 variants. In cells expressing A120 or T120 PAR4 proteins, no differences in half-maximal effective concentration in receptor activation by PAR4-AP were observed. BMS-986120 was well tolerated with dose-proportional pharmacokinetics and concentration-dependent pharmacodynamics in healthy participants over a wide dose range.
ClinicalTrials.gov ID: NCT02208882.
Introduction
Balance between antithrombotic efficacy and bleeding with currently available antiplatelet drugs is suboptimal. In patients at low risk for occlusive vascular events, benefit of acetylsalicylic acid (ASA) on reducing vascular events is offset by bleeding complications [Citation1–3]; addition of clopidogrel or other P2Y12 inhibitors to ASA in higher risk situations brings increased bleeding together with superior efficacy to ASA alone [Citation2]. These mechanism-based bleeding events have restricted the use of stronger antiplatelet agents to improve antithrombotic efficacy. Furthermore, in patients treated with ASA and P2Y12 inhibitors, pathologic thrombosis may occur via platelet activation by other platelet receptors, and many patients continue to experience recurrent ischemic events [Citation2]. There is a high unmet need for an agent with a novel mechanism that can reduce thrombotic events without increasing bleeding risk.
Thrombin, the most potent platelet activator, may play a major role in atherothrombosis. Thrombin activates human platelets via proteolytic cleavage of two distinct G-protein-coupled receptors called protease-activated receptor 1 and 4 (PAR1 and PAR4) [Citation4]. The high-affinity thrombin receptor PAR1 is the target of the antiplatelet agent vorapaxar, which has efficacy in reducing thrombotic events in patients with a history of myocardial infarction or with peripheral arterial disease [Citation5]. In patients with history of myocardial infarction, vorapaxar showed 20% reduction in the combined endpoint of cardiovascular death, myocardial infarction, or stroke compared with placebo [Citation6]. However, vorapaxar increased the risk of bleeding, including intracranial hemorrhage and fatal bleeding [Citation5,Citation6]. Vorapaxar, when added to standard of care in patients with acute coronary syndrome, was associated with increased bleeding risk while failing to provide additional benefit on the primary efficacy endpoint of recurrent ischemic cardiovascular events [Citation7]. To identify an agent that could further improve clinical outcome without added bleeding risk, we sought to target PAR4, a low-affinity thrombin receptor believed to mediate a late stage of platelet activation, contributing to occlusive thrombus formation but playing a lesser role in primary hemostasis [Citation8].
We recently reported discovery of an orally active, potent, and selective PAR4 antagonist, BMS-986120 [Citation9]. When evaluated in a cynomolgus monkey model of arterial thrombosis and provoked bleeding, BMS-986120 exhibited low bleeding liability and a markedly wider therapeutic window than the P2Y12 antagonist clopidogrel [Citation9]. Recently, BMS-986120 was administered to humans and reduced ex vivo platelet-rich thrombus formation under high shear stress [Citation10].
Platelets from individuals with PAR4 variant T120, which is more common in Black than White individuals (63% vs 19%), show hyperresponsiveness to PAR4-AP–induced aggregation compared with those carrying the A120 variant [Citation11,Citation12]. This common variant of PAR4 might contribute to the disparity in thrombotic risk between Black and White individuals, and impact the pharmacodynamics of PAR4 antagonists [Citation11]. Thus, it is of interest to investigate the outcomes of BMS-986120 in preclinical and clinical studies.
Herein, we report results of two phase I, randomized, double-blind, placebo-controlled studies in healthy participants—a single-ascending oral dose (SAD) and a multiple-ascending oral dose (MAD) study—investigating oral PAR4 antagonist BMS-986120. These studies assessed the safety, tolerability, pharmacokinetics, and pharmacodynamics of BMS-986120. Because preclinical data indicate that CYP3A4 is involved in metabolism of BMS-986120 (data unpublished), effect of BMS-986120 on midazolam pharmacokinetics was investigated. In a separate set of experiments, platelet responses to BMS-986120 in A120T variants were investigated in comparison to A120. The pharmacodynamic effects of BMS-986120 in these studies were measured using a novel PAR4-AP described in this issue of Platelets [Citation13].
Materials and methods
These studies were conducted in accordance with Good Clinical Practice and the Declaration of Helsinki. All participants provided informed written consent. The protocols, amendments, and informed consent received appropriate approval by the Institutional Review Board/Independent Ethics Committee (IntegReview, Austin, TX, USA) before study initiation. The clinical phase was conducted at PPD Development, LLC, Wilmington, NC, USA.
Study designs
The SAD study used a randomized, double-blind, placebo-controlled design. Within each dose panel, eight healthy participants were randomly assigned in a 3:1 ratio to receive a single dose of BMS-986120 (0.3, 1.0, 3.0, 10, 30, 75, and 180 mg) or matched placebo. In panels 1 (the initial dose panel) and 3 (the first panel with a predicted efficacious dose), a sentinel cohort of two participants (one active, one placebo) was randomized and dosed in a blinded fashion. After 3 days of monitoring for safety and tolerability, the remaining six participants were randomized (five active, one placebo) and dosed. Other dose panels did not include a sentinel cohort; all eight participants (six active, two placebo) were randomized to treatment.
The MAD study (NCT02208882) used a randomized, double-blind, placebo-controlled, sequential panel design. Within each dose panel (2.5, 10, 40, or 100 mg), eight participants were randomly assigned in a 3:1 ratio to receive once daily (QD) BMS-986120 or matched placebo for 14 days. Participants in the 40-mg dose panel also received a single oral 2.5-mg dose of midazolam, a sensitive CYP3A4 substrate, alone on day –1 (day before beginning BMS-986120 dosing) and coadministered with BMS-986120 or placebo on day 13 to assess whether BMS-986120 has the potential to induce CYP3A4.
Participants
Participants 18–65 years of age not of childbearing potential were eligible for the SAD study if they were considered healthy based on the absence of clinically significant deviations from normal in medical history, physical examination, 12-lead electrocardiogram (ECG), vital signs, clinical laboratory evaluations, and a body mass index of 18–32 kg/m2. Key inclusion criteria for the MAD study were the same as for the SAD study, except the age range was 18–75 years. Supplementary Materials show complete lists of exclusion criteria.
Rationale for dose selection
Doses of BMS-986120 investigated in the SAD study were selected to encompass the potential efficacious exposure range, while remaining at exposures deemed safe and tolerable based on nonclinical data for BMS-986120 [Citation9], ex vivo platelet inhibition [Citation10], allometric scaling for projected human pharmacokinetics, and clinical safety and pharmacokinetic data from initial dose panels. See Supplementary Materials for details.
Pharmacokinetics
In the SAD study, blood samples were taken predose and 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 9, 12, 24, 36, 48, 72, 96, 120, and 144 hours postdose for pharmacokinetic assessment.
In the MAD study, blood samples were taken on day 1 predose and 0.5, 1, 2, 3, 4, 6, 9, and 12 hours postdose; predose on days 2, 4, 7, 10, and 13; day 14 predose and at 0.5, 1, 2, 3, 4, 6, 9, 12, 24, 36, 48, 72, 96, 120, and 144 hours postdose. Additionally, in the 40-mg BMS-986120 plus 2.5-mg midazolam panel, plasma samples were collected predose and 0.25, 0.5, 1, 2, 3, 4, 6, 9, 12, and 24 hours after day –1 and day 13 dosing to assess whether BMS-986120 has the potential to induce CYP3A4. Supplementary Materials show details of sample collection and analysis for both studies.
Pharmacodynamics
Pharmacodynamic assessment included platelet aggregation. In the SAD study, samples were taken predose and 1, 2, 3, 4, 12, 24, 48, 72, 96, and 144 hours after dosing. In the MAD study, samples were taken on day 1 predose and 1, 2, 4, 9, and 12 hours postdose; predose on days 2, 4, 7, and 10; day 14 predose and 1, 2, 4, 9, 24, and 72 hours postdose.
Platelet aggregation was analyzed at the clinical site using a validated ex vivo assay performed on platelet-rich plasma, described in Yang et al. [Citation13]. A novel PAR4-AP (A-Phe[4-F]-PGWLVKNG) was used to induce platelet activation and aggregation [Citation13]. PAR4-AP and PAR1-AP (SFFLRR; GenScript, Piscataway, NJ, USA) were prepared as 20× stock solutions using Hanks’ Balanced Saline. Platelet aggregation was initiated by the addition of 25 µL of PAR4-AP or PAR1-AP stock solutions to 475 µL of platelet-rich plasma and monitored for 10 minutes using light transmission aggregometry instruments (Chrono-log Corporation, Havertown, PA, USA). Various concentrations of PAR4-AP (12.5, 25, 50, and 100 µM) were tested, and 25 µM PAR1-AP was used as a control.
Genotyping
PAR4 genotyping was performed to determine the A120T variant for each participant. See Supplementary Materials for details.
Statistical considerations
No statistical power calculations were used for either study. The planned administration of BMS-986120 to six participants per dose panel provided an 80% probability of observing at least one occurrence of any adverse event (AE) that would occur with 24% incidence in the population from which the sample was drawn. All statistical analyses were performed using SAS/STAT Version 9.2 (SAS Institute, Inc., Cary, NC, USA). See Supplementary Materials for details.
Safety
Safety was assessed using AEs, laboratory evaluations, ECGs, vital signs, and physical examinations. Recorded AEs were tabulated by treatment. Additional safety assessments in both studies included fecal occult blood and template bleeding time [Citation14].
In the MAD study, ECGs were performed at 0, 1, 2, 3, 4, 9, and 24 hours postdose on day 1. Time-matched ECGs were to occur within 15 minutes of baseline on day –1, and the 24-hour baseline ECG on day –1 was equivalent to the day 1 predose (0 hours) evaluation. Any abnormalities were listed after evaluation by the investigator. Vital signs were recorded on day 1, before dosing, and every 24 hours until study discharge. Serial measurements of seated blood pressure and resting heart rate were obtained immediately before dosing and 1, 2, 3, 4, 6, 9, 12, and 24 hours postdose. Time-matched baseline measurements of blood pressure and heart rate were obtained on day –1. The 24-hour baseline measurement was equivalent to the day 1 predose measurement.
Mutagenesis study methods
PAR4 expression and function was investigated using a stable HEK293 cell line expressing human PAR4, in which the thrombin cleavage site was modified by replacing Arg-47/Gly-48 with a 6x histidine tag. Site-directed mutagenesis was used to evaluate the effect of the A120T variant. PAR4-AP stimulated Ca2+ signaling was monitored using a fluorescent Ca2+-sensitive indicator. Surface expression of PAR4 was determined by FACS using an anti-His antibody. The specific maximal binding and the affinity of PAR4 for BMS-986120 were determined by radioligand binding studies using membranes prepared from HEK293 cells expressing PAR4 and a 3H-BMS-986120 ligand. Expression level at the cell surface of PAR4 was determined by FACS using an anti-His antibody and a goat anti-mouse-Oregon Green 488 as primary and secondary antibodies, respectively. See Supplementary Materials for details.
Immunohistological study methods
Preclinical research was conducted on donor tissue to investigate the expression of PAR1 and PAR4 in human central nervous system tissues susceptible to intracranial parenchymal hemorrhage. See Supplementary Materials for details.
Results
Participant disposition and baseline characteristics
A total of 56 healthy participants were randomized, treated, and analyzed in the SAD study. All 56 completed the study as planned. A total of 32 healthy participants were randomized in the MAD study; two participants (BMS-986120 2.5-mg [Citation1] and 100-mg [Citation1] groups) discontinued before study completion because of AEs considered unrelated to treatment. Baseline demographics and clinical characteristics are shown in Tables SI and SII. In the SAD study, the population was 76.8% male and predominantly White (67.9%) or Black (30.4%), with a mean age of 40.8 years. The MAD study population had a mean age of 37.1 years, and was 93.8% male, 68.8% White, and 21.9% Black.
Pharmacokinetics
BMS-986120 was readily absorbed after single-dose administration of 0.3–180 mg as an oral spray dried dispersion formulation (). Median time to maximum concentration (Tmax) ranged from 1 to 3 hours across all doses evaluated, and mean half-life was approximately 45–84 hours at the dose range of 3.0–180 mg. Exposure to BMS-986120 increased in an approximately dose-related manner after administration of single oral doses from 0.3 to 180 mg (). The point estimates and 90% confidence intervals (CIs) for the slope of the regression line (i.e. coefficient β in the relation of BMS-986120 pharmacokinetic parameter = A × Doseβ) were slightly above 1 for AUC from time zero extrapolated to infinite time (β = 1.119; 90% CI, 1.072–1.165) and AUC from zero to the time of the last quantifiable concentration (β = 1.100; 90% CI, 1.054–1.145), and slightly below 1 for maximum concentration (Cmax; β = 0.942; 90% CI, 0.908–0.977).
Figure 1. Plasma concentration‒time profiles of BMS-986120.
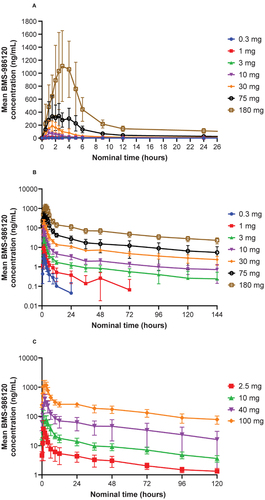
Table I. Pharmacokinetics.
BMS-986120 was readily absorbed after multiple-dose administration of 2.5–100 mg daily; median Tmax was 1.5 hours for each dose on day 1, and at day 14 was 2 hours for all doses except 10 mg (3 hours; ). The accumulation index suggests up to a twofold increase in AUC at steady state compared with day 1. Half-life ranged from 57.1 to 80.4 hours; the effective half-life, based upon the accumulation index of BMS-986120 ranged from 18.9 to 24.7 hours. The concentration–time curve after dosing on day 14 shows approximately dose-proportional exposure (), consistent with linearity assessments performed for both SAD and MAD data. The pharmacokinetics of midazolam, a sensitive 3A4 substrate, were similar on days –1 and 13, suggesting no notable interaction between BMS-986120 and midazolam (Figure S1).
Pharmacodynamics
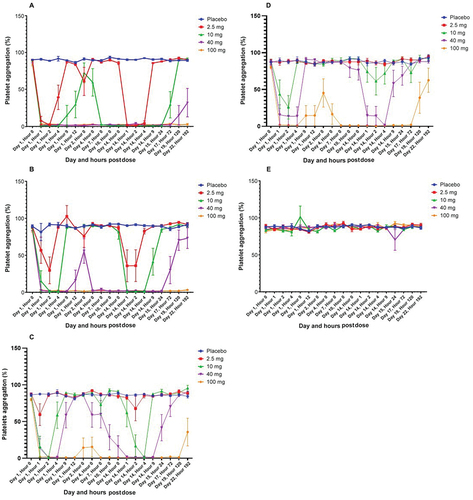
BMS-986120 selectively inhibited PAR4-AP-induced ex vivo platelet aggregation in a dose- and concentration-dependent manner. All four concentrations of PAR4-AP (12.5, 25, 50, and 100 μM) generated a maximal aggregation response before dosing. In the SAD study (Figure S2), the lowest dose of BMS-986120 showing evidence of a pharmacologic effect on aggregation induced by 12.5 μM PAR4-AP was 1.0 mg; only 3/6 participants in this dose panel achieved ≥80% maximal inhibition of PAR4-AP-induced platelet aggregation, and the effect was not sustained through 24 hours postdose in any participant. All participants in the 3.0-mg dose panel achieved ≥80% inhibition, but the inhibition was not sustained for 24 hours postdose. At least 80% inhibition through the 24-hour postdose interval was observed for 2/6 participants receiving 10 mg and 5/6 at a dose of 30 mg; all participants in the 75-mg and 180-mg groups maintained ≥80% inhibition through ≥24 hours postdose. At higher concentrations of PAR4-AP (25, 50, and/or 100 μM), the dose‒response curve was shifted to the right (i.e. higher doses of BMS-986120 were required to achieve ≥80% inhibition sustained for ≥24 hours). At the highest concentration of PAR4-AP (100 μM), a 10-mg dose yielded maximal but nonsustained inhibition of ≥80% in all or nearly all participants, and a single dose of 180 mg yielded sustained (induced platelet aggregation persisted for 24 hours) inhibition of ≥80% in three to five participants.
As plasma drug concentrations declined over time after dosing (), the extent of inhibition of platelet aggregation decreased, reflecting concentration responses to 12.5 and 25 µM PAR4-AP (); 12.5 and 25 µM PAR4-AP). A complete inhibition of platelet aggregation for at least 24 hours was observed in all participants with BMS-986120 doses ≥10 mg daily within an hour of first dose administration, and the extent of inhibition of platelet aggregation decreased as plasma drug concentrations declined (); 12.5 and 25 µM PAR4-AP). At the highest concentration of PAR4-AP (); 100 μM), sustained inhibition throughout the treatment period was achieved by BMS-986120 doses of 100 mg daily for 3/6 participants. As evidence of selectivity, PAR1-AP-induced platelet aggregation was unaffected ().
Figure 2. Induced platelet aggregation in the multiple-ascending dose (MAD) study.
Pharmacokinetic/pharmacodynamic relationship
Time-matched pharmacokinetic/pharmacodynamic data pooled from both SAD and MAD studies demonstrated dose- and concentration-dependent inhibition of PAR4-AP-induced platelet aggregation with dose-related onset and offset (). There was a steep pharmacokinetic/pharmacodynamic response across a range of PAR4-AP concentrations.
Figure 3. Platelet aggregation responses to PAR4-AP by self-identified race.
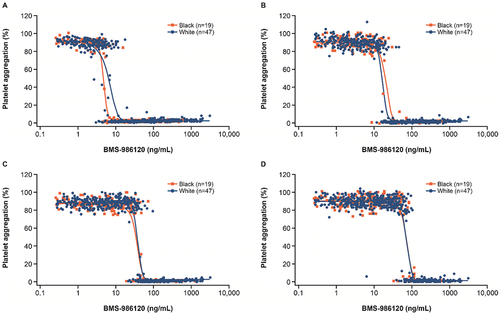
Figure 4. Inhibition of human platelet aggregation by BMS-986120 in participants sorted by PAR4 A120T variants.
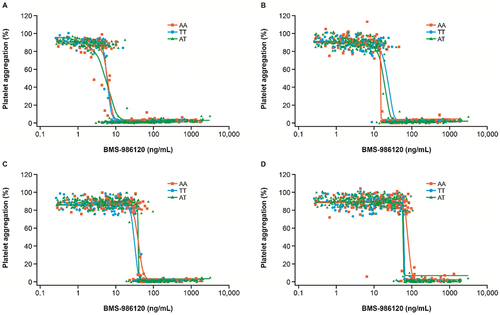
When pooling data from both SAD and MAD studies of BMS-986120, there was a steep concentration-response to platelet aggregations across a range of PAR4-AP concentrations regardless of self-identified race (). The “on/off” effect of inhibition of platelet aggregation appeared to increase a similar extent in groups self-identified as being Black or White with increasing doses of PAR4-AP. Furthermore, the BMS-986120 pharmacokinetic/pharmacodynamic relationship did not show differences in PAR4-mediated platelet response between participants carrying AA versus TT or A/T variants in pooled data from both studies ().
Safety
AEs for both studies are summarized in Table SIII and SIV. No bleeding-related AEs were reported in either study; there were no positive test results for fecal occult blood. In the SAD study, bleeding times across doses and times ranged from a low of 0.5 minutes with placebo and BMS-986120 3.0 mg to a high of 12 minutes with BMS-986120 1.0 mg and 75 mg; mean baseline values ranged from 3.08 to 5.08 minutes and the max increase from baseline was 7 minutes with BMS-986120 1.0 mg. In the MAD study, bleeding times across doses and times ranged from a low of 0.5 minutes with placebo to a high of 8 minutes with BMS-986120 40 mg plus midazolam and BMS-986120 10 mg. No deaths or severe AEs occurred.
In the SAD study, the most frequent AEs were headache (2 [4.8%] with BMS-986120) and dizziness (1 [2.4%] with BMS-986120 and 1 with [7.1%] placebo); these were the only events assessed as related to treatment. No participant discontinued because of AEs. Twenty-four participants had ECG abnormalities, including 11 with sinus bradycardia present after dosing; no ECG abnormalities were reported by the investigator as AEs. No participant receiving BMS-986120 had a QT interval corrected for heart rate via Fridericia’s method (QTcF) greater than 450 ms, and only one participant receiving BMS-986120 (180 mg) had a QTcF change from baseline >30 ms. This participant had a QTcF of 439 ms at day 1, hour 9, representing a 35-ms increase from the time-matched baseline value on day –1.
In the MAD study, one participant receiving placebo had a mild drug-related AE of diarrhea; no other AEs were deemed related to the study drug. Two participants discontinued because of AEs not related to the study drug (one nonserious, moderate case of nephrolithiasis 1 hour after administration of BMS-986120 2.5 mg QD on day 10, treated with hydrocodone, ibuprofen, and tamsulosin; one nonserious skin infection on the lower left leg approximately 23 hours after BMS-986120 100 mg QD, requiring incision and drainage of the infected area and treatment with doxycycline hyclate, lidocaine, bactroban (mupirocin), and betadine (povidone iodine). No bleeding time-related AEs were reported. There were ECG abnormalities reported at screening, day −1, and predose on day 1, though no ECG abnormalities were reported as AEs. No participant had a QTcF interval >450 ms; two participants had QTcF change from baseline >30 ms (40 and 41 ms).
Mutagenesis study results
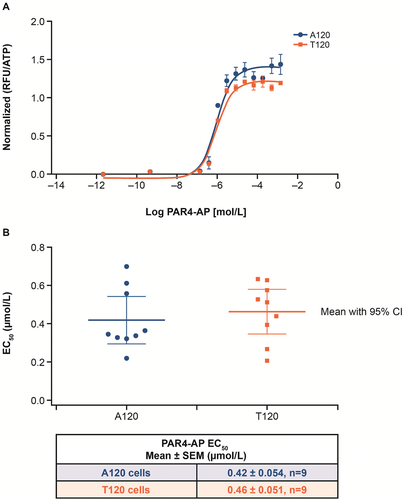
A side-by-side comparison of PAR4-AP-induced calcium mobilization assay results conducted using HEK293 cells expressing either A120 or T120 PAR4 protein variants demonstrated no differences in mean effective concentration values (). The minimal differences in the PAR4-AP maximal response between the two variants are consistent with the small differences in cell surface receptor levels (Figure S3).
Figure 5. Influence of A120T variants on PAR4-AP-induced calcium mobilization.
Discussion
BMS-986120 has dose-proportional pharmacokinetics and predictable, concentration-dependent pharmacodynamic responses on platelets based on findings from a SAD and MAD study. In the SAD study, all participants in the 75- and 180-mg groups maintained ≥80% inhibition through at least 24 hours postdose. In the MAD study, complete inhibition of platelet aggregation for at least 24 hours was observed in all participants with doses ≥10 mg daily after ~7 days. Reversibility of effect was demonstrated by return of platelet aggregation induced by 25 µM PAR4-AP to near baseline levels 24 hours after repeated dosing of 10 mg BMS-986120. The degree of inhibition was diminished when increasing concentrations of PAR4-AP were used to stimulate platelet activation, suggesting surmountable inhibition versus PAR4-AP. Among PAR4-AP concentrations, the antithrombotic efficacy of BMS-986120 in previous monkey studies corresponded best with the inhibition of platelet aggregation response to PAR4-AP at concentrations of 12.5 and 25 µM. Therefore, inhibition of platelet aggregation induced by 12.5 and 25 µM PAR4-AP is considered a suitable surrogate to assess the antiplatelet pharmacodynamic effect of BMS-986120.
Half-life in the SAD study displayed an apparent trend of increasing with dose (from 44.7 hours with 3.0 mg to 84.1 hours with 180 mg). However, this may be partly attributable to the limits of quantification, as suggested by the fact that half-life could not be determined for the 0.3- and 1.0-mg doses in this study, and no clear trend of longer half-life with increasing dose was observed after 14 days administration in the MAD study. Based on preliminary data from the SAD study, the estimated effective half-life and the superposition principle, BMS-986120 was expected to have a two-fold (100%) increase in AUC in 1 dosing interval and a 20% increase in Cmax on day 14, relative to exposures on day 1; this was largely borne out by the results of the MAD study, with the exception of a small decrease in Cmax in the 10-mg dose group. Given the effective half-life of approximately 20 hours, QD dosing was considered reasonable. In the MAD study, doses of ≥10 mg maintained through concentrations at or above the projected threshold concentration (range of 4–30 nM or 2–15 ng/mL) over a 24-hour dosing interval, and in line with projections for the “on/off” effect, maintained ≥80% inhibition of PAR4-AP-induced platelet aggregation over 24 hours after 14 days QD administration.
Our unpublishted data suggest that BMS-986120 may be metabolized by CYP3A4 and that BMS-986120 may be an inhibitor of CYP3A4. Reaction phenotyping studies with recombinant human CYP enzymes suggest that CYP3A4 is the primary enzyme responsible for the oxidative metabolism of BMS-986120. Further, in human liver microsomes, BMS-986120 is a time-dependent inhibitor of CYP3A4 (IC50 = 1.1–1.5 µM). Based on our preclinical data, the effect of BMS-986120 on midazolam pharmacokinetics was investigated. The notable lack of interaction between BMS-986120 and midazolam suggests BMS-986120 will not have significant effects on CYP3A4 enzymes. Furthermore, the non-linearity in the data is likely due to the poor solubility of BMS-986120 and its highly protein-bound state (>99.9%) in vitro in human serum.
As with the PAR1 antagonist vorapaxar [Citation15], BMS-986120 was not associated with relevant changes in bleeding time in healthy volunteers. Additionally, in cynomolgus monkeys, BMS-986120 had a markedly wider therapeutic window than clopidogrel [Citation9]. In the SAD and MAD studies, BMS-986120 was well tolerated and not associated with any bleeding AEs. Furthermore, preliminary immunohistochemical results indicate that PAR4 does not appear to be expressed in human brain tissues, whereas PAR1 expression was detected in the cerebral microvasculature, predominantly in the tunica adventitia, with increased levels observed in and around stroke/hemorrhage lesions (Figure S4). This is consistent with the finding that no PAR4 expression was detected in human brain tissue [Citation16]. Although it is not clear if inhibition of PAR1 in the brain vasculature would manifest as intracranial bleeding, nonplatelet-related bleeding is evident in PAR1-deficient mice [Citation17]. Because PAR1 is not expressed on normal murine platelets, the observed bleeding is explained by a known endothelial-specific function of PAR1 in maintaining vascular integrity. The lack of substantial PAR4 expression in the human brain might mitigate the potential risk of bleeding associated with PAR4 inhibition. The safety profile of BMS-986120 in this phase I study is consistent with findings from our preclinical study in cynomolgus monkeys [Citation9].
In this study, the variability seen in template bleeding times in the single-ascending dose study post dosing was assessed as not drug-dependent or clinically relevant by the investigators and may be due to technical variability. Additionally, the maximum mean ± SD increase from baseline in template bleeding times, across all BMS-986120 doses and time points, was 1.58 ± 0.25 minutes compared with the placebo mean baseline range of 3.08–5.08 minutes. There were no clinically relevant mean changes in any hematology, serum chemistry, and coagulation parameter, and no participant had a positive fecal occult test. While it is possible that combination of BMS-986120 with other bleeding-time prolonging compounds that synergize with PAR4 inhibition may prolong bleeding times in an additive or synergistic manner, this effect was not tested here. Therefore, it would have to be explicitly tested in future studies.
In a separate report focusing on an unselected general population, the authors found that abnormal ECG findings (the most prevalent being bundle branch blocks and hemiblocks) are highly common in the general population aged 40 years or older, with only about half the participants evaluated having completely normal ECGs [Citation18]. Furthermore, in another separate report evaluating results from four large Belgian epidemiological studies, the authors found that ECG abnormalities in the adult population are not rare, and the study found no dose- or concentration-related trends for any ECG parameter, diastolic or systolic blood pressure, or heart rate following administration of BMS-986120 [Citation19]. Some participants had ECG abnormalities at screening or predose, underscoring the benign nature of these common ECG abnormalities (Supplemental Table 4). ECG abnormalities observed in this study were not considered AEs related to the study drug as assessed by the investigators and are not reported in the Supplemental Table 3 (Adverse Events).
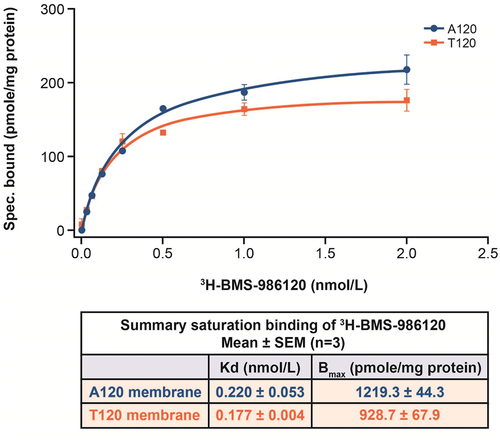
The potential clinical utility of PAR4 antagonism is given important context by the investigations into the effect of race and genetic variations presented here. Research by Edelstein et al. found that the PAR4 genetic variant T120 is more common in the Black population than in the White population, and it has been suggested that this T/A dimorphism at residue 120 contributes to a subtle difference in response to PAR4-AP and can affect the in vitro response to PAR4 antagonism [Citation11]. However, the degree of variability seen in platelet response according to genotype varied as a function of agonist concentration used, in that the published variant effect is subtle and detected only at submaximal concentrations of PAR4-AP [Citation11,Citation20]. Recent work found that platelets carrying the AA120 variant required twice the concentration of PAR4-AP AYPGKF for irreversible aggregation [Citation21]. Additionally, AA120 platelets required a threefold higher PAR4-AP AYPGKF concentration for PAR4 desensitization [Citation12]. Our clinical results for BMS-986120 do not suggest a potential differential effect by allele or race. The phase I study described here used an optimized PAR4-AP [Citation13] at full response, but the concentration response in vitro in the mutagenesis study did not support a difference even at lower PAR4-AP concentrations. The marginal differences in the maximal response observed between cells expressing the A120 versus T120 variants can be explained by the slightly higher level of receptor expressed at the cell surface for the A120 variant (). These findings do not support dose adjustment for BMS-986120 based on single nucleotide polymorphisms at residue 120 of PAR4. This is broadly consistent with results of an ex vivo study in 530 individuals that found no significant shift in potency or antiplatelet effect of the small-molecule PAR4 antagonist UDM-002555 according to genetic variants [Citation22].
Figure 6. Saturation binding of 3H-BMS-986120.
Limitations of this study include the fact that, despite the pooling of data from both SAD and MAD studies for analysis, the evaluation of differential response by racial/genetic factors was based on a small sample size. The point estimates and 90% CIs for the slope of the regression lines for pharmacokinetic parameters were slightly above or below 1; however, these results should be interpreted with caution because this study was not designed or powered to confirm meaningful departures from dose proportionality. It is also challenging to find bleeding events in phase 1 studies, even in compounds with liability for bleeding events. Finally, these studies in healthy volunteers may not be representative of the effects of BMS-986120 in patients with cardiovascular diseases who would be likely to receive antithrombotic therapy.
These results represent the first reported clinical experience of a selective oral PAR4 antagonist. BMS-986120 was safe and well tolerated in these healthy participants and had predictable pharmacokinetics/pharmacodynamics, suggesting that PAR4 antagonist therapy may have promising potential in patients at thrombotic risk. In healthy human participants, the pharmacodynamic effect of the PAR4 antagonist did not appear to differ in participants carrying AA or TT variants on residue 120 of PAR4.
Author contributions
MB and MC conducted the research.
MC generated figures.
All authors contributed to, read, edited, and approved the manuscript.
Supplemental Material
Download PDF (418.9 KB)Acknowledgements
The authors thank Jacques Banville, Julia Guy, E. Scott Priestley, Anne Marinier, and Roger Rémillard (design and synthesis of BMS-986120); Anh Bui, Itay Perlstein, Joanne Ma, Amber Griffies, and Cynthia Yones (execution of the clinical studies); Jocelyne Guay, Jeffrey Colin, and Sophie Desmeules (mutagenesis study: in vitro calcium and radioligand binding assays); Asterand Bioscience and Jian Chen (brain histology work); Erin Siegfried (genotyping); Fraz Ahmed Ismat (development of the manuscript). Professional medical writing and editorial assistance provided by Rob Coover, MPH, and Nicole Draghi, PhD, at Caudex, and Scott Forbes, PhD, at Oxford PharmaGenesis Inc. Medical writing was funded by Bristol Myers Squibb.
Disclosure statement
S. Merali, Z. Wang, C. Frost, M. Hedrick, L. Hui, S. Meadows Shropshire, and K. Xu are employees of Bristol Myers Squibb. M.M. DeSouza and J. Yang are former employees of Bristol Myers Squibb.
M. Callejo and M. Bouvier are employees of the Institute for Research in Immunology and Cancer at the Université de Montréal and participated in this study in the context of a collaboration with Bristol Myers Squibb. M. Bouvier holds a Canada Research Chair in Signal Transduction and Molecular Pharmacology.
Data availability statement
Requests to access the dataset from qualified researchers trained in human participant confidentiality protocols may be made via Bristol Myers Squibb’s online request system at https://fasttrack-bms.force.com/.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/09537104.2022.2088719
Additional information
Funding
References
- Coccheri S. Use and misuse of aspirin in primary cardiovascular prevention. Clin Med Insights Cardiol 2017; 11: 1179546817702149. Epub 2017/05/05. doi:10.1177/1179546817702149
- Eikelboom JW, Hirsh J, Spencer FA, Baglin TP, Weitz JI. Antiplatelet drugs: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2012; 141: e89S–e119S. Epub 2012/02/15. doi:10.1378/chest.11-2293
- Whitlock EP, Burda BU, Williams SB, Guirguis-Blake JM, Evans CV. Bleeding risks with aspirin use for primary prevention in adults: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med 2016; 164: 826–835. Epub 2016/04/12. doi:10.7326/M15-2112
- Heuberger DM, Schuepbach RA. Protease-activated receptors (PARs): mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb J 2019; 17: 4. Epub 2019/04/13. doi:10.1186/s12959-019-0194-8
- Zontivity® (vorapaxar) [Prescribing information]. 2014. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204886s000lbl.pdf
- Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, Fox KA, Lipka LJ, Liu X, Nicolau JC, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med 2012; 366: 1404–1413. Epub 2012/03/27. doi:10.1056/NEJMoa1200933
- Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van de Werf F, White HD, Aylward PE, Wallentin L, Chen E, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med 2012; 366: 20–33. Epub 2011/11/15. doi:10.1056/NEJMoa1109719
- Holinstat M, Voss B, Bilodeau ML, McLaughlin JN, Cleator J, Hamm HE. PAR4, but not PAR1, signals human platelet aggregation via Ca2+ mobilization and synergistic P2Y12 receptor activation. J Biol Chem 2006; 281: 26665–26674. Epub 2006/07/14. doi:10.1074/jbc.M602174200
- Wong PC, Seiffert D, Bird JE, Watson CA, Bostwick JS, Giancarli M, Allegretto N, Hua J, Harden D, Guay J, et al. Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci Transl Med 2017; 9. Epub 2017/01/06. doi:10.1126/scitranslmed.aaf5294
- Wilson SJ, Ismat FA, Wang Z, Cerra M, Narayan H, Raftis J, Gray TJ, Connell S, Garonzik S, Ma X, et al. PAR4 (protease-activated receptor 4) antagonism with BMS-986120 inhibits human ex vivo thrombus formation. Arterioscler Thromb Vasc Biol 2018; 38: 448–456. Epub 2017/12/23. doi:10.1161/ATVBAHA.117.310104
- Edelstein LC, Simon LM, Lindsay CR, Kong X, Teruel-Montoya R, Tourdot BE, Chen ES, Ma L, Coughlin S, Nieman M, et al. Common variants in the human platelet PAR4 thrombin receptor alter platelet function and differ by race. Blood 2014; 124: 3450–3458. Epub 2014/10/09. doi:10.1182/blood-2014-04-572479
- Whitley MJ, Henke DM, Ghazi A, Nieman M, Stoller M, Simon LM, Chen E, Vesci J, Holinstat M, McKenzie SE, et al. The protease-activated receptor 4 Ala120Thr variant alters platelet responsiveness to low-dose thrombin and protease-activated receptor 4 desensitization, and is blocked by non-competitive P2Y12 inhibition. J Thromb Haemost 2018; 16: 2501–2514. Epub 2018/10/23. doi:10.1111/jth.14318
- Yang J, Mapelli C, Wang Z, Sum CS, Hua J, Lawrence RM, Ni Y, Seiffert DA. An optimized agonist peptide of protease-activated receptor 4 and its use in a validated platelet-aggregation assay. Platelets 2022; 1–8. Epub 2022/03/29. doi:10.1080/09537104.2022.2053091
- Chen F, Maridakis V, O’Neill EA, Beals C, Radziszewski W, de Lepeleire I, Van Dyck K, Depre M, Bolognese JA, de Hoon J, et al. A randomized clinical trial comparing point-of-care platelet function assays and bleeding time in healthy subjects treated with aspirin or clopidogrel. Platelets 2012; 23: 249–258. Epub 2011/09/17. doi:10.3109/09537104.2011.604806
- Kraft WK, Gilmartin JH, Chappell DL, Gheyas F, Walker BM, Nagalla S, Naik UP, Horrow JC, Wrishko RE, Zhang S, et al. Effect of vorapaxar alone and in combination with aspirin on bleeding time and platelet aggregation in healthy adult subjects. Clin Transl Sci 2016; 9: 221–227. Epub 2016/06/16. doi:10.1111/cts.12405
- Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW, Foster DC. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A 1998; 95: 6642–6646. Epub 1998/06/17. doi:10.1073/pnas.95.12.6642
- Griffin CT, Srinivasan Y, Zheng YW, Huang W, Coughlin SR. A role for thrombin receptor signaling in endothelial cells during embryonic development. Science 2001; 293: 1666–1670. Epub 2001/09/05. doi:10.1126/science.1061259.
- Awamleh Garcia P, Alonso Martin JJ, Jimenez Hernandez RM, Graupner Abad C, Talavera Calle P, Serrano Antolin J, Cristobal Varela C, Curcio Ruigomez A, Muniz J, Gomez Doblas JJ, et al. Abnormal electrocardiographic findings in the population older than 40 years. Prevalence and clinical significance. Results of the OFRECE study. Rev Esp Cardiol (Engl Ed) 2019; 72: 820–826. Epub 2019/02/19. doi:10.1016/j.rec.2019.01.001
- De Bacquer D, De Backer G, Kornitzer M. Prevalences of ECG findings in large population based samples of men and women. Heart 2000; 84: 625–633. Epub 2000/11/18. doi:10.1136/heart.84.6.625
- Edelstein LC, Simon LM, Montoya RT, Holinstat M, Chen ES, Bergeron A, Kong X, Nagalla S, Mohandas N, Cohen DE, et al. Racial differences in human platelet PAR4 reactivity reflect expression of PCTP and miR-376c. Nat Med 2013; 19: 1609–1616. Epub 2013/11/13. doi:10.1038/nm.3385
- Morikawa Y, Kato H, Kashiwagi H, Nishiura N, Akuta K, Honda S, Kanakura Y, Tomiyama Y. Protease-activated receptor-4 (PAR4) variant influences on platelet reactivity induced by PAR4-activating peptide through altered Ca2+ mobilization and ERK phosphorylation in healthy Japanese subjects. Thromb Res 2018; 162: 44–52. Epub 2018/01/01. doi:10.1016/j.thromres.2017.12.014
- Downes K, Astle W, McKinney H, Batista J, Kempster C, Garner S, Allegretto N, Ouwehand WH, Yang J. Platelet responsiveness to PAR4 activation in a healthy population and its inhibition by a potent and selective PAR4 antagonist. Eur Heart J 2016; 37(suppl 1): 1017.