
Abstract
The association between T-cell large granular lymphocytes (T-LGL) and ITP is uncertain. The aims of this study were to determine the prevalence of T-LGL in patients with ITP and to describe its association with ITP disease severity. We analyzed flow cytometry results for T-LGL (using a threshold of 0.3 x109 or greater cells/L) or positive T-cell receptor clonality in patients with ITP and nonimmune thrombocytopenia. Descriptive statistics were used to characterize the association between T-LGL and ITP, response to ITP treatments (rituximab and splenectomy) and response to T-LGL treatment. Among ITP patients, 14.3% (13/91) had evidence of a T-LGL population compared to 10.3% (3/29) of patients with non-immune thrombocytopenia. ITP patients with T-LGL had lower nadir platelet counts (2 vs. 47 × 109/L) and received more ITP treatments (median 6 vs. 3) than ITP patients without T-LGL. Response to rituximab was observed in 14.3% (1/7) of ITP patients with T-LGL and 54.5% (6/11) without T-LGL. Response to splenectomy was observed in 25% (2/8) with T-LGL and 56.2% (9/16) without T-LGL. Four patients with ITP and T-LGL received treatment for T-LGL with methotrexate; none had an improvement in platelet count levels. T-LGL may appear in patients with ITP, and the meaning of this finding remains unclear; however, for some patients, the presence of abnormal T-LGL may indicate a more severe form of ITP that tends to be less responsive to therapy. In this cohort, treatment of T-LGL with methotrexate did not improve platelet counts in the few patients who were treated.
Introduction
Large granular lymphocytes (LGL) represent 10% to 15% of total peripheral blood mononuclear cells in adults and consist of T-cell and natural killer (NK) subsets [Citation1]. A proliferation of T-cell large granular lymphocyte (T-LGL) cells can occur as a reactive process in the setting of autoimmune disease, viral infections, connective tissue disease, skin disorders and non-Hodgkin lymphoma [Citation2,Citation3] or as a malignant process [Citation4,Citation5]. T-LGL cells are CD4- and CD8+, with the characteristic phenotypic markers CD3+, CD16+, CD57+ [Citation6,Citation7]. Clonal populations of T-LGL are characterized by a T-cell receptor (TCR) gene rearrangement. LGL malignancies constitute 2% to 5% of lymphoproliferative disorders including T-LGL leukemia, which tends to run an indolent course [Citation1,Citation8]. Previous criteria from the 2008 World Health Organization classification defined T-LGL leukemia as an increase of T-LGL cells (>2 x109/L) for at least 6 months without a clear cause; however, it is now recognized that a lower cell count may be compatible with the diagnosis [Citation1,Citation8]. The diagnosis of T-LGL leukemia requires evidence of LGL on a peripheral blood smear and a restricted LGL clone identified by TCR gene rearrangement in the setting of cytopenias, lymphocytosis, autoimmune conditions, splenomegaly and/or recurrent infection [Citation4,Citation8]. Methotrexate is considered first-line therapy for indolent LGL leukemia [Citation7].
Excess T-LGL cells or evidence of TCR clonality can occur more broadly in the setting of autoimmune diseases, specifically rheumatoid arthritis (RA), pure red cell aplasia, autoimmune hemolytic anemia, and Evans syndrome even without meeting criteria for T-LGL leukemia. The association between T-LGL and immune thrombocytopenia (ITP) has rarely been reported [Citation6,Citation8–10]. ITP is an autoimmune disease characterized by a platelet count below 100 × 109/L and an increased risk of bleeding. Primary ITP occurs in the absence of an underlying cause, but secondary forms of ITP can occur in the setting of infection, drugs, autoimmune diseases or lymphoproliferative diseases [Citation11]. Whether T-LGL should be considered among the secondary causes of ITP is uncertain [Citation12]. In this study, we examined the prevalence of excess T-LGL cells in patients with ITP and described the association between T-LGL and ITP disease outcomes.
Methods
Prospective cohort study of consecutive patients with ITP and thrombocytopenia due to nonimmune causes.
Population
Patients were identified from the McMaster ITP Registry, an ongoing, prospective registry of consecutive adult patients with thrombocytopenia (platelet count <150 x109/L) who were referred to a tertiary hematology clinic at McMaster University Medical Centre in Hamilton, Canada. Enrollment began in 4 January 2010. All patients were treated as per clinical need and no intervention was applied. Follow-up visits occurred every six or 12 months until discharge from clinic or death. Peripheral blood flow cytometry to identify T-LGL was done routinely for all patients as of 2018. Prior to that date, the test was performed at the discretion of the physician. For this study, we included adults ≥18 years old enrolled in the registry up to 31 December 2019. A dedicated research coordinator collected clinical data, laboratory results, and bleeding assessments using the ITP Bleeding Scale [Citation13]. The diagnosis of ITP was established by two hematologists working in the clinic and was based on criteria put forth by the American Society of Hematology [Citation11]. The registry also included patients with thrombocytopenia that was due to nonimmune causes, such as liver disease, hypersplenism, and myelodysplasia. Those patients were used as controls for this study. Response to rituximab or splenectomy was defined as an increase in the platelet count level to 50 × 109/L or higher and doubling of baseline within 12 weeks of the treatment.
T-LGL
All patients who had peripheral blood leukocyte phenotyping by flow cytometry after enrollment into the registry or up to 6 months before enrollment in the registry were included. For the flow cytometry technique, we used a whole blood lysis method. Three washes are done on specimens: two washes with Phosphate Buffered Saline (PBS) with 1% Bovine Serum Albumin (BSA), one wash with Roswell Park Memorial Institute (RPMI) medium and 10% Fetal Calf Serum (FCS). The antibodies are Duraclone pre-mixed, dried down antibodies. The washed specimen is added and incubated for 15 min in the dark at room temperature. The specimen is lysed with in-house made ammonium chloride. The specimen is washed a further 2 times in PBS BSA then data is acquired (more details in the Supplemental Material). Testing for T-LGL was done for patients who had CD3+CD56+ cells >6% of the total leukocyte population, or CD4:CD8 ratio <0.5, or if specifically requested by the treating physician. We defined T-LGL as CD3+ CD16+CD57+ lymphocytes, and we used evidence of clonality to distinguish between reactive forms and more aggressive form of T-LGL [Citation14]. Clonal T-LGL tends to present with CD4 or CD8 restriction, express pan-T cell antigens, and show TCR gene rearrangement gamma delta or alpha beta restriction. TCR gene rearrangement was determined by polymerase-chain reaction (PCR) for patients with T-LGL >2 x109/L or if specifically requested. Excess T-LGL was defined as total T-LGL cell count >0.3 x109/L [Citation8] or evidence of clonality by TCR gene rearrangement by PCR.
Statistical analysis
Categorical data were reported as frequencies and proportions, and continuous variables were reported as means and standard deviations (SDs), or medians with interquartile ranges (IQRs) if data were skewed. Fisher exact test was used to analyze the significance of differences in proportions.
Results
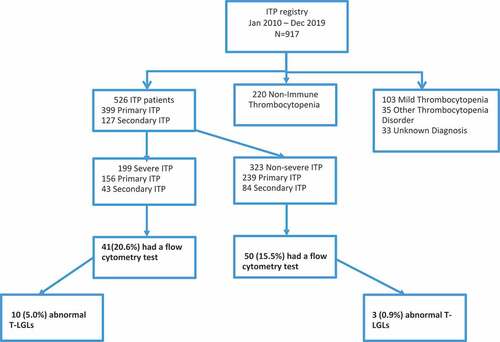
From 4 January 2010 to 31 December 2019, 917 patients were enrolled in the McMaster ITP Registry and of those, 269 were screened for T-LGL by flow cytometry. Fourteen patients were excluded because they had mild thrombocytopenia only (platelet count 100–150 x109/L), eight patients were excluded because they had heparin-induced thrombocytopenia or thrombotic thrombocytopenic purpura, and 11 patients were excluded because the diagnosis of the thrombocytopenia could not be resolved. Among the remaining 236 patients (158 with ITP and 78 with nonimmune thrombocytopenia), 205 (86.9%) had flow cytometry test results available either on peripheral blood or bone marrow samples. Patients with bone marrow samples only (n = 85) were excluded because the criteria for excess T-LGL >0.3x109/L could only be applied to peripheral blood samples and imputation was not possible. Thus, 91 patients with ITP (69 primary ITP and 22 secondary ITP) and 29 patients with non-immune thrombocytopenia were included in the analysis. We analyzed the group of patients who were recruited after 2018 (n = 103) separately since this cohort had flow cytometry testing done routinely for all consecutive patients. This group included 78 patients with ITP (62 primary ITP and 16 secondary ITP) and 25 patients with non-immune thrombocytopenia.
T-LGL among ITP patients and thrombocytopenic controls
We collected 139 flow cytometry tests (n = 120 patients); 120 tests (86.3%) were done as part of screening or reflex testing, and 19 (13.7%) were specifically requested by the physician. In the ITP group (n = 91), 13 (14.3%) had T-LGL. Baseline patient characteristics for patients with immune thrombocytopenia (ITP) with and without abnormal T-LGL are shown in . Flow cytometry phenotyping for the 13 patients with ITP and T-LGL is shown in . Of the 13 patients, 11 had excess T-LGL (>0.3 x109/L) and two had positive TCR clonality only. Median T-LGL cell count per patient was 0.5 x109/L (IQR 0.4–1.3) and none had >2.0 x109/L cells. All 10 patients who were tested had evidence of a clonal TCR gene rearrangement. In the nonimmune thrombocytopenia group (n = 29), three (10.3%) had T-LGL. The median T-LGL cell count for the control group was 1.0 x109/L (IQR 0.3–1.6). One patient was tested for TCR gene rearrangement and the result showed a clonal population. There was no significant difference in the proportion of patients with ITP and/or non-immune thrombocytopenic patients with T-LGL (p = .23). All patients with ITP and T-LGL belong to group of 78 ITP patients who had flow cytometry done after 2018. Only one patient with non-immune thrombocytopenia and T-LGL had the flow cytometry done before 2018.
Table I. Baseline patient characteristics for patients with immune thrombocytopenia (ITP) with and without abnormal T-large granular lymphocytes (T-LGL).
Table II. Phenotype of ITP patients with T-LGL.
Clinical characteristics of ITP patients
Among the 13 patients with T-LGL-positive ITP, 10 (76.9%) had severe ITP (nadir platelet below 30 × 109/L) and the remaining 3 (23.1%) patients were mild (). The median platelet count at the time of T-LGL testing for patients with T-LGL-positive ITP was lower [16 x 109/L (IQR 5–35) vs. 74 × 109/L (IQR 34–122)] and median nadir platelet counts was lower [(2 x 109/L (IQR 1–19) vs. 47 × 109/L (IQR 13–85)] compared with T-LGL-negative ITP patients. T-LGL-positive ITP patients had a median of three major bleeds and received a median of six ITP treatments compared with one major bleed and three prior treatments for the T-LGL-negative ITP group.
Figure 1. Selection of patients.
Seven T-LGL-positive patients and 11 T-LGL-negative patients received Rituximab as a treatment for ITP. One of the 7 ITP patients (14.3%) with T-LGL achieved a transient response to rituximab compared to 6 of 11 (54.5%) ITP patients without T-LGL, including 4 who were receiving other concomitant ITP treatments. Eight T-LGL-positive patients and 16 T-LGL-negative patients had a splenectomy for their ITP. Two of 8 (25%) T-LGL-positive patients achieved a platelet count response (of those, one was also receiving other concomitant treatment for ITP), compared with 9 of 16 T-LGL-negative patients (56.2%).
We identified four patients with T-LGL-positive ITP who were treated with methotrexate as a potential treatment for secondary ITP due to T-LGL (). These patients had received a median of seven prior treatments for ITP and had a median platelet count of 34 × 109/L prior to the initiation of methotrexate. The median duration of methotrexate treatment was 5.5 months. None of the patients had a platelet count response after methotrexate. For three patients, flow cytometry was repeated after treatment and one patient had an improvement in the number of T-LGL cells.
Table III. Characteristics of patients with T-LGL who were treated with methotrexate (MTX).
Discussion
In this study, we aimed to establish the prevalence of excess or clonal T-LGL cells in patients with ITP and whether this finding has any clinical associations with ITP outcomes. There was no significant difference in the prevalence of T-LGL among patients with ITP (15.4%) compared with patients with thrombocytopenia due to nonimmune causes (10.3%) (p = .1620). Nevertheless, patients with T-LGL-positive ITP appeared to have more severe disease. Only four patients received methotrexate with the hopes of reducing the T-LGL population and improving the thrombocytopenia; however, while the number of T-LGL cells was reduced in one patient, none had an improvement in platelet count levels.
Clonal or oligoclonal T-LGL expansion has been described in patients who may be minimally symptomatic and do not meet criteria for LGL leukemia including patients with autoimmune diseases. The term T-cell clonopathy of unknown or undetermined significance or T-CUS has been applied in previous publications to distinguish this abnormal laboratory finding from T-LGL leukemia, analogous to monoclonal gammopathy of undetermined significance [Citation4,Citation15]. The importance of identifying clonal T-cell expansions has been demonstrated in patients with autoimmune diseases, such as rheumatoid arthritis, since long-term exposure to tumor necrosis factor – inhibitors as a treatment may be linked to a greater risk of clonal T-cell expansions, which in turn may cause severe cytopenias [Citation9]. A previous study described seven patients with chronic ITP and concurrent indolent T-cell clonopathy, all of whom were refractory to more than three ITP treatments, but with anecdotal response to azathioprine, supporting the concept of cytotoxic T-lymphocytes as a possible pathogenic mechanism of the chronic ITP [Citation16]. A recent study found an increased cytotoxic potential of CD8+ T cells in a subset of ITP patients compared to healthy controls [Citation17]; however, evidence for platelet-specific CD8+ T cells is lacking [Citation18]. The presence of CD56+ T-LGL cells has been reported to indicate an aggressive clinical course, but this is not always the case [Citation19]. In our population, 11 of the 13 patients with T-LGL had CD56+, and none had an aggressive form of T-LGL. TCR clonality has been associated with a platelet count response after treatment with rituximab in a previous study of ITP patients (n = 30) [Citation12]. Mouse models have shown that anti-CD20 therapy can reduce the ability of CD8(+) T cells to cause thrombocytopenia [Citation20]. Our data suggested that the presence of T-LGL might reduce the likelihood of a treatment response with rituximab; however, the number of observed patients was small.
Strengths of this study were the collection of data from a registry designed specifically to study ITP and other thrombocytopenia disorders, rather than hospital-based or administrative databases. All patient samples were tested by flow cytometry in the same reference laboratory using the same testing technique and criteria for interpretation. The use of nonimmune thrombocytopenic controls provided a meaningful comparator to identify specific characteristics of patients with ITP that may contribute to the disease. Previous ITP studies that have used non-immune thrombocytopenia as controls have yielded informative results [Citation21–23]. Long follow-up time and duplicate assessment of diagnosis were also strengths.
Limitations were the retrospective design and missing TCR data on some patients. The T-LGL ITP group has three patients without TCR gene rearrangement by PCR data which can influence the results in this small dataset (n = 13), however one patient had other markers suggestive of a clonal T-LGL population (CD2+, dim CD5+, CD4-, CD8- and TCR-gamma-delta+). Collection of samples for flow cytometry testing was not done at standardized points in time for patients enrolled in the registry before 2018, however the majority of tests in this study (86.3%) was routinely performed at the time of enrollment (after 2018), thus minimizing sampling bias. T-LGL occurrence appeared to be associated with time from diagnosis, which may be a confounder. We used previously established criteria to identify T-LGL (absolute count >0.3x109/L as a criteria for T-LGL overall; and other markers suggestive of clonal changes or absolute count >2 x109/L as a cutoff for testing for TCR gene rearrangement) [Citation1,Citation5,Citation7,Citation8]. Different treatments for ITP may have altered the results of lymphocyte phenotyping by flow cytometry, so it is possible that we underestimated the frequency of LGL since most all patients had received ITP treatment before LGL was assessed by flow, and treatment may have cleared an abnormal LGL population. Our patient population derived from a tertiary hematology referral center, which may have resulted in selection bias and over-representation of patients with more severe ITP. The small sample size limits the inferences that can be made from this study.
In conclusion, our study showed that T-LGL abnormalities are not more frequent in patients with ITP than in patients with nonimmune thrombocytopenia. However, ITP patients with concomitant T-LGL abnormalities appeared to have a more severe form of ITP. Treatment with methotrexate did not improve platelet counts in the four patients with refractory ITP. Further clinical and biochemical studies are needed to understand the effect of abnormal lymphocytes on the development of thrombocytopenia and on ITP outcomes. Our results do not justify routine testing for T-LGL for all patients with ITP.
Authorship contributions
C.G. helped design the study, collected the data, wrote the manuscript, and approved the final version; Y.L. helped design the study, performed the statistical analysis, edited the manuscript and approved the final version; J.D.; M.S.J. and K.J.L. helped collect the data, edited the manuscript, and approved the final version; D.K. helped perform the experiments, edited the manuscript, and approved the final version; J.G.K. conceived the study, edited the manuscript, and approved the final version; and D.M.A. conceived the study, wrote the manuscript, and approved the final version.
Supplemental Material
Download PDF (340.6 KB)Acknowledgments
The disclosure statement has been inserted. Please correct if this is inaccurate.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/09537104.2022.2144194.
Additional information
Funding
References
- Sokol L, Loughran TP Jr. Large granular lymphocyte leukemia. Oncologist. 2006;11(3):263–5. doi:10.1634/theoncologist.11-3-263.
- Loughran TP Jr. Clonal diseases of large granular lymphocytes. Blood. 1993;82(1):1–14. doi:10.1182/blood.V82.1.1.bloodjournal8211.
- Okuno SH, Tefferi A, Hanson CA, Katzmann JA, Li CY, Witzig TE. Spectrum of diseases associated with increased proportions or absolute numbers of peripheral blood natural killer cells. Br J Haematol. 1996;93(4):810–812.
- Sabnani I, Tsang P. Are clonal T-cell large granular lymphocytes to blame for unexplained haematological abnormalities? Br J Haematol. 2007;136(1):30–37. doi:10.1111/j.1365-2141.2006.06374.x.
- Oshimi K. Clinical features, pathogenesis, and treatment of large granular lymphocyte leukemias. Intern Med. 2017;56(14):1759–1769. doi:10.2169/internalmedicine.56.8881.
- Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist. 2004;9(3):247–258. doi:10.1634/theoncologist.9-3-247.
- Lamy T, Loughran TP Jr. How I treat LGL leukemia [published correction appears in blood. 2014 Mar 27;123(13):1182]. Blood. 2011;117(10):2764–2774. doi:10.1182/blood-2010-07-296962.
- Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood. 2017;129(9):1082–1094. doi:10.1182/blood-2016-08-692590.
- Schwaneck EC, Renner R, Junker L, Einsele H, Gadeholt O, Geissinger E, Kleinert S, Gernert M, Tony H-P, Schmalzing M. Prevalence and characteristics of persistent clonal T cell large granular lymphocyte expansions in rheumatoid arthritis: a comprehensive analysis of 529 patients. Arthritis Rheumatol. 2018;70(12):1914–1922. doi:10.1002/art.40654.
- Zhang R, Shah MV, Loughran TP Jr. The root of many evils: indolent large granular lymphocyte leukaemia and associated disorders. Hematol Oncol. 2010;28(3):105–117. doi:10.1002/hon.917.
- Neunert C, Terrell DR, Arnold DM, Buchanan G, Cines DB, Cooper N, Cuker A, Despotovic JM, George JN, Grace RF, et al. American society of hematology 2019 guidelines for immune thrombocytopenia [published correction appears in blood adv. 2020 Jan 28;4(2):252]. Blood Adv. 2019;3(23):3829–3866. doi:10.1182/bloodadvances.2019000966.
- Stasi R, Del Poeta G, Stipa E, Evangelista ML, Trawinska MM, Cooper N, Amadori S. Response to B-cell–depleting therapy with rituximab reverts the abnormalities of T-cell subsets in patients with idiopathic thrombocytopenic purpura. Blood. 2007;110(8):2924–2930. doi:10.1182/blood-2007-02-068999.
- Page LK, Psaila B, Provan D, Michael Hamilton J, Jenkins JM, Elish AS, Lesser ML, Bussel JB. The immune thrombocytopenic purpura (ITP) bleeding score: assessment of bleeding in patients with ITP. Br J Haematol. 2007;138(2):245–248. doi:10.1111/j.1365-2141.2007.06635.x.
- Moignet A, Lamy T. Latest advances in the diagnosis and treatment of large granular lymphocytic leukemia. Am Soc Clin Oncol Educ Book. 2018;38(38):616–625. doi:10.1200/EDBK_200689.
- Dhodapkar MV, Li CY, Lust JA, Tefferi A, Phyliky RL. Clinical spectrum of clonal proliferations of T-large granular lymphocytes: a T-cell clonopathy of undetermined significance? Blood. 1994;84(5):1620–1627. doi:10.1182/blood.V84.5.1620.1620.
- Sabnani I, Tsang P. Therapeutic implications of T-cell clonopathy of unknown significance in chronic immune thrombocytopenic purpura. Platelets. 2009;20(2):135–139. doi:10.1080/09537100802657727.
- Vrbensky JR, Arnold DM, Kelton JG, Smith JW, Jaffer AM, Larché M, Clare R, Ivetic N, Nazy I. Increased cytotoxic potential of CD8+ T cells in immune thrombocytopenia. Br J Haematol. 2020;188(5):e72–76. doi:10.1111/bjh.16334.
- Vrbensky JR, Nazy I, Clare R, Larché M, Arnold DM. T cell-mediated autoimmunity in immune thrombocytopenia. Eur J Haematol. 2022;108(1):18–27. doi:10.1111/ejh.13705.
- Alekshun TJ, Tao J, Sokol L. Aggressive T-cell large granular lymphocyte leukemia: a case report and review of the literature. Am J Hematol. 2007;82(6):481–485. doi:10.1002/ajh.20853.
- Guo L, Kapur R, Aslam R, Speck ER, Zufferey A, Zhao Y, Kim M, Lazarus AH, Ni H, Semple JW. CD20+ B-cell depletion therapy suppresses murine CD8+ T-cell–mediated immune thrombocytopenia. Blood. 2016;127(6):735–738. doi:10.1182/blood-2015-06-655126.
- Li N, Heddle NM, Nazy I, Kelton JG, Arnold DM. Platelet variability index: a measure of platelet count fluctuations in patients with immune thrombocytopenia. Blood Adv. 2021;5(20):4256–4264. doi:10.1182/bloodadvances.2020004162.
- Vrbensky JR, Nazy I, Toltl LJ, Ross C, Ivetic N, Smith JW, Kelton JG, Arnold DM. Megakaryocyte apoptosis in immune thrombocytopenia. Platelets. 2018;29(7):729–732. doi:10.1080/09537104.2018.1475637.
- Nazy I, Kelton JG, Moore JC, Clare R, Horsewood P, Smith JW, Ivetic N, D’Souza V, Li N, Arnold DM. Autoantibodies to thrombopoietin and the thrombopoietin receptor in patients with immune thrombocytopenia. Br J Haematol. 2018;181(2):234–241. doi:10.1111/bjh.15165.