
Abstract
High-altitude polycythemia (HAPC) can occur in individuals who are intolerant to high-altitude hypoxia. In patients with HAPC, erythrocytosis is often accompanied by a decrease in platelet count. Chronic hypoxia can increase the incidence of arteriovenous thrombosis and the risk of bleeding during antithrombotic treatment due to thrombocytopenia; therefore, understanding the cause of thrombocytopenia can reduce the risk of treatment-related bleeding. In this study, we examined platelet production and apoptosis to understand the cause of thrombocytopenia in patients with HAPC. The classification of myeloid-derived megakaryocytes (MKs) in HAPC patients was mainly granular MKs rather than mature MKs, suggesting impaired differentiation and maturation. However, the total number of MKs and newly generated reticulated platelets in the peripheral blood increased, indicating sufficient platelet generation in HAPC thrombocytopenia. Increased platelet apoptosis may be one of the causes of thrombocytopenia. Platelet activation and GP1bα pathway activation induced by thrombin and von Willebrand factor can lead to platelet apoptosis. Platelet production was not reduced in patients with HAPC, whereas platelet apoptosis was associated with thrombocytopenia. These findings provide a rationale for considering the bleeding risk in HAPC patient while treating thrombotic diseases.
Plain Language Summary
What is the context?
Platelets are essential in the process of blood clotting; hence, low platelet count increases the risk of bleeding. Thrombocytopenia is present in patients with high-altitude polycythemia
Hypoxia can lead to platelet activation and increase in procoagulant factors, while at the same time increase the risk of thrombosis due to erythrocytosis and blood stasis.
Antithrombotic therapy should be administered when thrombosis occurs in patients with high altitude polycythemia; however, due to the low platelet count, risk of bleeding must be considered.
What is new?
In this study, we found that platelet production was not decreased in patients with high-altitude polycythemia.
One cause of thrombocytopenia is apoptosis, which is associated with platelet activation, especially GP1bα activation.
Inhibition of GP1bα binding to ligand decreased the level of platelet apoptosis.
What is the impact?
This study provides novel insights into antithrombotic therapy for patients with high-altitude polycythemia complicated by thrombosis.
Thrombocytopenia is associated with excessive apoptosis.
Interfering with GP1bα targets may have a dual benefit, both in inhibiting thrombosis and avoiding thrombocytopenia.
Introduction
There are 140 million people living in the highlands around the world [Citation1]. People who are intolerant to hypoxia may suffer from chronic mountain sickness that is characterized by high-altitude polycythemia (HAPC; females Hb 19 g/dL; males Hb 21 g/dL), with an incidence of 5%–10% [Citation2]. Hypoxic environments inhabited by patients with HAPC transform blood into a hypercoagulable state, thereby increasing the risk of arteriovenous thrombosis [Citation3–7]. Antithrombotic therapy is the primary treatment option for these patients, but caution is warranted in some cases, including thrombocytopenia. A meta-analysis of the effects of high altitude exposure on platelet counts (PCs) showed that acute hypoxia had no significant effect on PCs, but chronic hypoxia, especially in patients with high altitude polycythemia, had a significant decrease in PCs [Citation8]. Therefore, understanding thrombocytopenia can lead to improved treatment outcomes when using antithrombotic drugs. If platelet output is reduced, anticoagulation and antiplatelet therapy should be used with caution, as patients may experience excessive bleeding. There are several pathological states, including disseminated intravascular coagulation, sepsis, and rheumatoid arthritis, in which PC also decreases. These effects are partly related to excessive platelet activation and subsequent platelet consumption [Citation9,Citation10]. Procoagulant platelets, which are essential for clot formation, can undergo apoptosis or necrosis [Citation11–13]. If thrombocytopenia in HAPC patients is closely related to platelet activation, anticoagulation and antiplatelet therapy should be sufficient to inhibit thrombosis and relieve thrombocytopenia.
Therefore, in this study, we investigated platelet generation and clearance in patients with HAPC to provide treatment guidance to better mitigate thrombocytopenia and thrombosis.
Methods
Collection of clinical data
All study subjects provided informed consent, and the study protocol was approved by the Qinghai University Affiliated Hospital ethics committee (P-SL-2020087). All subjects were male Han Chinese from Qinghai Province, China (2,270–4,500 m). Males are reportedly more prone to HAPC than females [Citation14,Citation15]. To exclude a gender difference, we recruited only males. Blood samples were collected on the day of admission from HAPC patients (HAPC group; n = 73) who were hospitalized in the Qinghai University Affiliated Hospital from 1 January 2017, to 31 May 2021. The reasons for hospitalization in HAPC group were secondary symptoms of polycythemia caused by long-term high altitude hypoxia, including headache, dizziness, asthma, palpitations, fatigue, etc. The diagnostic criteria for HAPC are according to the Qinghai Standard of Chronic Mountain Sickness (CMS), formulated by the sixth international conference on high altitude medicine and hypoxia physiology in 2004 [Citation2]. Specifically, it refers to the clinical syndrome caused by gradual mal-acclimatization to the hypoxic environment of the plateau in people who have lived on the plateau above 2500 meters for a long time (≥ 5 years). The main manifestation is erythrocytosis, in which the hemoglobin of males is ≥ 21 g/dL. The following conditions should be excluded, including patients with hypoxemia and secondary polycythemia due to chronic lung disease, chronic respiratory disorders or other diseases, as well as patients with polycythemia vera (AK2/V617F mutation testing was not performed on the HAPC cohort; polycythemia vera was excluded based on the fact that all HAPC participants had a long history of residing on a plateau, during which the number of neutrophils and platelets in their peripheral blood did not increase). Simultaneously, blood samples were collected from healthy subjects who arrived from high-altitude areas (2,500–4,500 m; HA group; n = 73) and moderate-altitude areas (1,500–2,500 m; MA group; n = 79) in the same hospital during the same period. Healthy subjects were hospitalized patients who intended to be admitted for removal of internal fixation at least 1 year after internal fixation. Subjects with tumors, hypertension, diabetes, coronary heart disease, rheumatic diseases, thrombosis, and hemorrhagic disorders, as well as those who had taken any anti-platelet or anti-thrombotic medications during the previous month, were excluded based on medical records and physical examination data. The blood samples were examined using an automatic hematology analyzer (SYSMexXE-2100; Sysmex, Hyogo, Japan).
Bone marrow smear by bone marrow aspirate were collected from patients with HAPC (HAPC group; n = 55) and from patients with leukemia who had achieved complete remission more than 2 years after chemotherapy and whose bone marrow smears were taken more than 3 months after the last chemotherapy (control group; n = 25). All participants were hospitalized in Qinghai University Affiliated Hospital from 1 January 2017, to 31 May 2021, and came from high-altitude areas (2,500–4,500 m). All megakaryocytes (MKs) were counted on films from a 1.5 × 3.0-cm bone marrow smear stained with Wright’s stain and classified as megakaryoblast (stage I), promegakaryocyte (stage II), granular MK (stage III), or mature MK (stage IV) [Citation16].
As the number of stage I and II MKs in the bone marrow smear appeared to be low and indistinguishable under a light microscope, these MKs were combined into one group. The proportions of various types of MKs were compared between the HAPC and control groups.
Antibodies and reagents
The reagents and kits used in this study were as follows: prostaglandin E1 (Cayman Chemical, Ann Arbor, MI, USA); a MicroBeads kit (Miltenyi Biotec, Gladbach Bergisch, Germany); human thrombin protein (G-CLONE, Beijing, China); human von Willebrand factor protein (Sino Biological, Beijing, China); ristocetin (HYPHEN BioMED, Neuville-Sur-Oise, France); fluorescein isothiocyanate (FITC)-conjugated Annexin V apoptosis detection kit and flow cytometry mitochondrial membrane potential detection kit (BD Biosciences, Franklin Lakes, NJ, USA); phycoerythrin (PE)-conjugated anti-human CD62P (P-selectin), PE-conjugated mouse anti-human IgG isotype, and PE-conjugated anti-human CD42b/glycoprotein (GP)Ibα (BioLegend, San Diego, CA, USA); MitoProbe tetramethylrhodamine methyl ester (TMRM) kit for flow cytometry (Invitrogen, Carlsbad, CA, USA); Thiazole Orange dye (Solarbio, Beijing, China); anti-caspase-3 and human CD42b/GPIbα enzyme-linked immunosorbent assay (ELISA) kit (NOVUS Biologicals, Littleton, CO, USA); rabbit polyclonal anti-CD42b (Proteintech, Rosemont, IL, USA); human vWF ELISA kit (Abcam, Cambridge, UK); thrombin/antithrombin complex ELISA kit (Cloud-Clone Corp, Katy, TX, USA); human C–X–C motif chemokine ligand 4 (CXCL4) ELISA Kit (BOSTER, Wuhan, China).
Platelet isolation
Antecubital venous blood was collected from subjects in HAPC group and HA group using an 18 G needle, with the first 2 mL of blood discarded, the tourniquet loosened, and the subsequent 8 mL of blood retained in an ACD Vacutainer (BD Biosciences). Platelet-rich plasma (PRP) was isolated via whole-blood centrifugation at 100 × g for 20 min. The PRP was then centrifuged at 200 × g for 20 min in the presence of 100 nM prostaglandin E1 (PGE1, to prevent exogenous platelet activation) to obtain washed platelets. Purified platelets were obtained through negative selection of the washed platelets supplemented with PGE1 using immunomagnetic beads (anti-CD45 microbeads, which for depletion of leukocytes), as previously reported [Citation17–19]. Finally, the purity of the isolated platelets was confirmed to be > 99.9% by observation under the microscope. The supernatant from the PRP after removing the platelets was referred to as “platelet-poor plasma” and stored at −80°C. Purified platelets were resuspended in phosphate-buffered saline (PBS) for subsequent experiments.
Flow cytometry
Flow cytometry was used to measure the level of platelet phosphatidylserine (PS) externalization, mitochondrial membrane potential (MMP), P-selectin, presentation of GPIbα (CD42b) on the platelet membrane surface, and proportion of reticulated platelets. To evaluate PS levels, platelets (1 × 106) were diluted with 100 μL of 1× binding buffer from the apoptosis kit, followed by addition of 5 μL of FITC-conjugated Annexin V and incubation at 22°C for 15 min. We then added 400 μL of 1× binding buffer, and the results were detected within 1 h. To evaluate P-selectin levels, platelets (1 × 106) were diluted with 100 μL PBS, followed by the addition of 5 μL PE-conjugated mouse anti-human P-selectin and incubation at 22°C for 15 min. The platelets were then washed twice, and results were detected within 1 h, with PE-conjugated mouse anti-human IgG used as an isotype control. We then determined the MMP using JC-1. Platelets (1 × 106) were diluted with 500 μL working solution from the mitochondrial membrane potential detection kit, incubated at 37°C in the dark for 30 min, and washed twice, with the results detected within 1 h. To examine the mitochondrial transmembrane potential using TMRM, platelets (1 × 106) were diluted with 100 μL PBS, followed by addition of 1 μL TMRM from the MitoProbe TMRM kit and incubation at 37°C for 30 min in the dark. The platelets were washed twice, and results were detected within 1 h. To detect surface GPIbα (CD42b) levels on the platelet membrane, platelets (1 × 106) were diluted with 100 μL PBS, followed by the addition of 5 μL PE-conjugated mouse anti-human CD42b and incubation at 22°C for 15 min. The platelets were then washed twice, and results were detected within 1 h, with PE-conjugated mouse anti-human IgG used as an isotype control. To determine the ratio of reticulated platelets, platelets (1 × 106) were diluted with 1 mL of Thiazole Orange staining solution (final concentration: 0.04 μM), incubated at 22°C for 30 min, and washed twice. The results were detected within 1 h. The flow cytometers were BD FACSCelesta and BD FACSAria III. It must be pointed out that all the data of each experiment were completed on one flow cytometer. The analysis software is Flow Jo 9.0.
Western blot
Radioimmunoprecipitation assay lysis buffer (100 μL; containing phenylmethylsulfonyl fluoride) was added to 1 × 108 purified platelets and immediately stored at −80°C. Protein was quantified to ensure that the total protein load of each sample was 15 μg. We then used human polyclonal anti-CD42b and human anti-caspase-3 for quantitative detection. Proteins were separated using 10% (for CD42b) and 12% (for caspase-3) sodium dodecyl sulfate polyacrylamide gel electrophoresis, followed by transfer onto a polyvinylidene fluoride membrane, which was blocked with 5% bovine serum albumin (BSA; Solarbio) diluted with 1× Tris-buffered saline containing 0.5% Tween 20 (TBST) for 1.5 h. Both primary antibodies were diluted at 1:1,000 with 5% BSA and incubated with the membrane overnight at 4°C, after which the secondary antibody (1:1,000 dilution with 1× TBST) was added and incubated at 22°C for 1.5 h. The results were analyzed using ImageJ software (v1.8.0; NIH, Bethesda, MD, USA).
ELISA
Recovered platelet-poor plasma was thawed to 22°C and analyzed to detect platelet factor 4 (PF4), thrombin, vWF, and soluble GPIbα levels according to manufacturer instructions for each respective kit.
Statistical analysis
Normal distribution measurement data were expressed as mean ± standard deviation, whereas the skewed distribution measurement data were expressed as median. Groups were compared using independent t tests, paired t tests, one-way analysis of variance, or nonparametric tests. Correlations were determined using Spearman correlation analysis. Statistical analysis was performed using GraphPad Prism software [v8.0.2 (263); GraphPad Software, La Jolla, CA, USA).
Results
PCs in patients with HAPC decreased and were negatively correlated with hemoglobin concentration, hematocrit, and mean platelet volume (MPV)
We retrospectively analyzed blood samples from 73 patients diagnosed with HAPC (HAPC group), 73 healthy high-altitude subjects (HA group), and 79 healthy moderate-altitude subjects (MA group). The mean PC decreased (137.99 ± 42.96; P < .001), the proportion of thrombocytopenia (PC < 100 × 109/L) increased (16/73; P < .001), and the percent of platelet count below 75 × 109/L reached 9.59% in patients with HAPC ( and ). The analysis of a relationship between red cell parameters and PC revealed a significant negative correlation between PC and hemoglobin levels in the HAPC group (r = 0.323; P = .005), with the same trend observed between platelet and hematocrit volumes (r = 0.343; P = .003) ().
Figure 1. PC of patients with HAPC decreased and was negatively correlated with hemoglobin concentration, hematocrit, and MPV. (A and B) Comparison of PC and thrombocytopenia among the three groups. (C) Correlation between PC and hemoglobin concentration in the HAPC group. (D) Correlation between PC and hematocrit in the HAPC group. (E) Correlation between PC and MPV in all three groups. (F) Correlation between PC and MPV in the HAPC group. (G) Correlation between PC and altitude in the HA group.
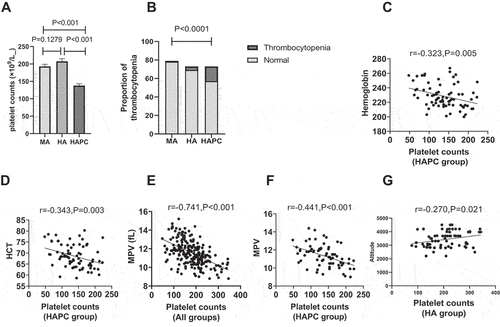
Table I. Hematological profiles and clinical characteristics among HAPC, HA, and MA groups.
The MPV indicates the size of platelets in the blood. Immature or newly formed platelets typically have higher volume [Citation20–23]. Thus, MPV can represent the platelet-producing activity of MKs in the bone marrow [Citation24]. In the present study, we observed a significant negative correlation between PCs and MPV not only in the HAPC group (r = 0.441; P < .001) but also in all groups (r = 0.741; P < .001) (), suggesting that peripheral thrombocytopenia stimulates hematopoietic tissue to produce new platelets. Comparison of the three groups showed that the number of platelets was lowest in the HAPC group while the MPV was not the highest (). These data suggest that the HAPC group demonstrated deficiency in producing new platelets. The correlation analysis between platelet count and altitude in all groups showed no correlation, and the same result was found in the HAPC group, but there was a low negative correlation in the HA group (r = 0.27, p = .021) (). This indicates that hemoglobin and hematocrit are more closely related to platelet count for patients with HAPC, and the effect of altitude on them is not obvious.
Patients with HAPC displayed deficiencies in differentiation and maturation during MK development but increases in newly generated platelets due to the increased MK count
The ability of hematopoietic tissue to produce platelets is related to the total number and type of MKs. The data demonstrated that the total number of MKs in the HAPC group increased significantly (63 (26, 143) vs. 29 (18, 70); P = .0235) (). Granular MKs (stage III) were predominant in the HAPC group (49.78 ± 16.12; P = .030), whereas mature MKs (stage IV) were predominant in the control group (46.16 ± 14.90; P = .003) (). This indicated that MKs from the HAPC group showed deficiencies in differentiation and maturation. Our previous research showed that platelets from healthy men exposed to hypoxia for 2 months presented a decreasing trend [Citation16]. Compared with the platelet proteomics of healthy controls, MK differentiation was negatively regulated as a biological process according to Gene Ontology analysis [Citation16], which is consistent with the present findings. Given the difficulty in collecting bone marrow samples, MK differentiation, apoptosis, and proliferation levels were not analyzed in the two groups. For further understanding platelet-production capacity under hypoxic conditions, we detected newly generated platelets by determining the ratio of reticulated platelets between the two groups.
Figure 2. Total number of MKs and reticulated platelets increased in the HAPC group. Bone marrow slices were obtained from patients with HAPC (HAPC group; n = 55), as well as from cured patients with non-megakaryocytic leukemia (control group; n = 25). (A and B) Comparison of the MK count and the classification proportions of MKs in bone marrow between the HAPC and control groups. (C) Representative figures of bone marrow slices stained using Wright’s stain. Granular MKs (stage III) and mature MKs (stage IV) are 40–70 μm in diameter, sometimes up to 100 μm. They have irregular morphology, large nuclei, irregular morphology, no nucleoli, and extremely abundant cytoplasm with a large number of small purply-red particles in the cytoplasm. The difference is that the granular MKs membrane is intact, and there is no platelet formation around the cells. However, the membrane of mature MKs is not clear, and most of them have pseudopodia, and platelets often accumulate on the medial and lateral sides. Granular MKs are indicated with red circles, and mature MKs are indicated with white circles. (D) Representative flow cytometric results of reticulated platelets. (E) Purified platelets were obtained and stained with Thiazole Orange to measure the reticulated platelet ratio using flow cytometry (n = 5 subjects/group).
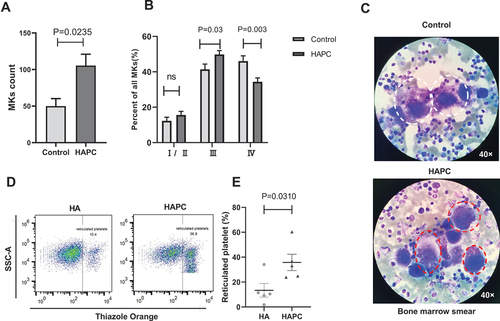
Reticulated platelets recently released from MKs reflect the platelet-producing capacity of MKs in hematopoietic tissue [Citation25–27]. In the present study, the ratio of reticulated platelets in the circulation of patients with HAPC was higher than that in the HA group (13.37 ± 12.19 vs. 35.76 ± 14.78; P = .0310) (). These results suggest that although MKs in the HAPC group demonstrated deficiencies in differentiation and maturation, the number of MKs increased along with the total number of platelets produced by hematopoietic tissue. The increased ratio of reticulated platelets in circulation is associated with arterial thrombosis, cardiovascular events, and venous thrombosis [Citation28–31]. Therefore, this might contribute to the high incidence of arteriovenous thrombosis in patients with HAPC. Since PCs in patients with HAPC were decreased while the number of newly generated platelets increased, we investigated platelet apoptosis.
Platelet apoptosis increased in patients with HAPC
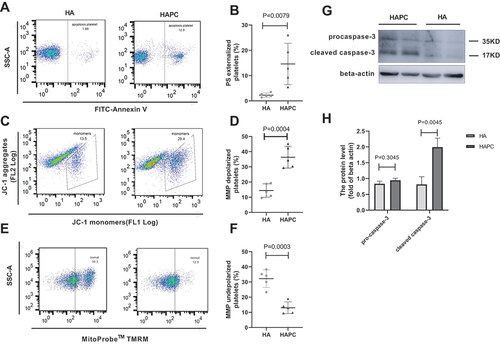
Apoptosis plays a key role in the clearance of platelets from circulation, primarily via the mitochondrial pathway [Citation32]. Specifically, binding of Bak/Bax to the mitochondrial membrane induces MMP depolarization and caspase-3 activation, which eventually result in platelet apoptosis [Citation32–35]. Since PS externalization is a marker of platelet apoptosis [Citation32,Citation36], we used FITC-labeled Annexin V to detect surface expression of PS on the platelet membrane. We found that PS externalization increased significantly (14.60 ± 8.17 vs. 2.28 ± 0.83; P = .0079) (), and that the MMP (according to JC-1 assays) in platelets decreased in the HAPC group relative to the HA group (36.30 ± 7.12 vs. 14.39 ± 4.44; P = .0004), suggesting increased platelet depolarization (). Moreover, western blot results showed that cleaved caspase-3 levels increased (1.61 (1.13, 3.08) vs. 0.45 (0.18, 1.47); P = .0045) (). These data indicated that platelet apoptosis increased in patients with HAPC, which may have caused thrombocytopenia. The HA group had a higher proportion of MMP-depolarized platelets compared to control groups in previous reports. We used JC-1 and TMRM methods to detect MMP, and both results showed the same trend. One study showed that hypoxia could cause a significant decrease in MMP [Citation37], so the reason for the high level of MMP depolarization in the HA group may be related to hypoxia due to a high-altitude lifestyle.
Figure 3. PS and caspase-3 levels increased and MMP decreased in the HAPC group. Flow cytometric detection of PS on and MMP in purified platelets (n = 5 subjects/group). Western blot detection of caspase-3 levels (n = 4–5 subjects). (A) Representative flow cytometric results for determining the presence of PS on the platelet surface labeled with FITC-conjugated Annexin V. (B) Comparison of PS levels between HAPC and HA groups. (C) Representative flow cytometric results of changes in platelet MMP using JC-1 (JC-1 monomers reflect MMP depolarization, and aggregates represent normal MMP). (D) Comparison of MMP using JC-1 between HAPC and HA groups. (E) Representative flow cytometric results of changes in platelet MMP using TMRM. Decreased fluorescence values indicate MMP depolarization. Scattered points in the box indicate platelets that have not undergone depolarization. (F) Comparison of MMP status using TMRM between HAPC and HA groups. (G, H) Caspase-3 levels according to western blot.
Platelet activation cause platelet apoptosis in patients with HAPC
Our previous research found that prolonged exposure to a hypobaric hypoxic environment increased platelet activation in humans [Citation19], which agreed with other studies on human subjects [Citation38,Citation39]. Additionally, previous studies showed that thrombin-activated platelets induce post-transcriptional regulation of Bcl-2 family proteins and activate caspases [Citation40]. Patients with HAPC were exposed to a hypoxic environment, and polycythemia can exacerbate hypoxia, which can increase thrombin levels and platelet activation.
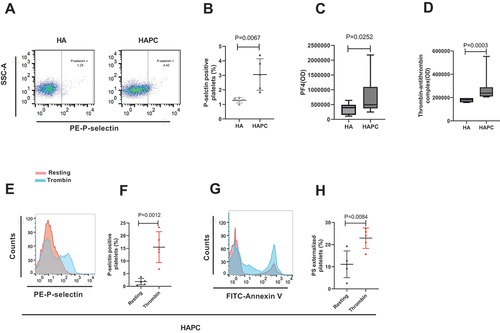
In this study, we detected levels of thrombin-antithrombin complex (which reflects the level of thrombin) and PF4 in plasma and P-selectin on the surface of platelet membranes from patients with HAPC. PF4 and P-selectin are specific proteins released by α granules during platelet activation and, thus, represent markers of platelet activation [Citation41,Citation42]. The results showed increased levels of plasma PF4 (492614 (371548, 1095700) vs. 397255 (151636, 491584); P = .0252), membrane P-selectin (3.06 ± 1.08 vs. 1.28 ± 0.18; P = .0067), and thrombin-antithrombin complex in plasma (237941 (209705, 288676) vs. 181176 (160000, 195882); P = .0003) in the HAPC group (). Notably, after stimulation with thrombin, P-selectin in patients with HAPC increased further (15.46 ± 6.11 vs. 1.86 ± 1.28; P = .0012) (), as well as PS externalization (22.96 ± 4.64 vs. 11.12 ± 6.06; P = .0084) (). This suggests that the increased baseline platelet activation in HAPC (increased P-selectin and PS) is driven by higher circulating thrombin levels, the mechanism of which remains unclear.
Figure 4. Platelet activation and apoptosis increased in the HAPC group. Platelets and plasma were isolated from HAPC and HA groups, and flow cytometry was used to detect PS and P-selectin on purified platelets (n = 5 subjects/group). (A) Representative flow cytometric results for P-selectin presentation on the platelet surface labeled with PE-conjugated CD62P. (B) Comparison of P-selectin levels between HAPC and HA groups. (C) Circulating plasma levels of PF4 measured using ELISA (HAPC: n = 23; and HA: n = 16). (D) Circulating plasma levels of thrombin measured by ELISA (HAPC: n = 8; and HA: n = 7). Human thrombin stimulates platelet activation. Purified platelets were incubated with 0.2 U/mL human thrombin at 22°C for 5 min. (E–G) Changes in (E, F) P-selectin and (G, H) PS expression before and after platelet activation.
The GPIb-IX-V signaling pathway is important for platelet clearance in patients with HAPC
GPIb-IX-V is a polymeric transmembrane platelet-receptor complex, and GPIbα is the primary subunit that binds to all known ligands of GPIb-IX-V, including vWF and thrombin [Citation43,Citation44]. The interaction between GPIbα and vWF is responsible for platelet adhesion to vascular damage [Citation45–50], with various studies showing that complex formation between platelet GPIbα and vWF is involved in platelet apoptosis and clearance. For example, interaction between GPIbα and vWF leads to translocation and cross-linking of the GPIb-IX-V complex in lipid rafts [Citation51–54]. Additionally, N-acetylglucosamine (GlcNAc), which prevents GPIbα translocation, inhibits platelet MMP depolarization and PS externalization and attenuates platelet apoptosis [Citation55].
Our previous research found a significant increase in transcript levels of VWF and GPIBA in human platelets exposed to hypoxia for more than 2 months [Citation19]. In the present study, we determined whether thrombocytopenia in patients with HAPC is also related to GPIb-IX-V signaling. First, we detected levels of plasma vWF and GPIbα (CD42b) on the platelet membrane in the HAPC group and found that plasma vWF increased (22.96 ± 4.64 vs. 11.12 ± 6.06; P < .0001) (), but there was a trend of reduction in percentage of GPIbα on platelet membranes (69.34 ± 20.32 vs. 92.24 ± 40.63; P = .0551) (). The primary pathological change in patients with HAPC was hypoxia. Then we wonder whether protein translation decreased or whether GPIbα had been shed from the membrane surface. The results revealed increases in both platelet (0.96 ± 0.15 vs. 0.65 ± 0.17; P < .0001) and plasma (178.27 ± 64.11 vs. 116.42 ± 71.08; P = .0356) levels of GPIbα (), suggesting that the ectodomain of GPIbα was shed. Ectodomain shedding of GPIbα has been observed in multiple studies of stored platelets and is related to platelet clearance, with inhibition of shedding capable of improving platelet-storage time [Citation56–58]. In the present study, we observed increased GPIbα shedding in the HAPC group, thereby providing novel insight into the underlying mechanism of thrombocytopenia in patients with HAPC.
Figure 5. Binding of vWF and GPIbα promotes platelet apoptosis and is accompanied by GPIbα shedding in the HAPC group. Platelets and plasma were isolated from HAPC and HA groups, and flow cytometry was used to detect PS and GPIbα (Cd42b) on purified platelets (n = 5/group). (A) Circulating plasma vWF levels were measured using ELISA (n = 12/group). (B) Representative flow cytometric results of CD42b presentation on the platelet surface labeled with PE-conjugated CD42b. (C, D) Comparison of CD42b percentage and MFI between HAPC and HA groups. (E) Circulating plasma levels of CD42b were measured using ELISA (n = 12/group). (F, G) CD42b levels in platelets according to western blot. Human thrombin, vWF, and ristocetin stimulate purified platelets. (H, I) Purified platelets from HAPC group were incubated with human thrombin (0.2 U/mL) at 22°C for 5 min, ristostine (0.5 mg/mL) and vWF (25 μg/mL) were then co-administered and incubated at 37°C for 15 min. Comparison of CD42b percentage and MFI before and after stimulation with thrombin, as well as VWF and ristocetin in the HAPC group. Platelet stimulation was inhibited via the GPIbα pathway using GlcNac. Purified platelets were incubated with GlcNac (100 mM) at 37°C for 30 min to inhibit GPIb-IX-V signaling, followed by subsequent incubation with ristocetin (0.5 mg/mL)+vwf (25 μg/mL) at 37°C for 15 min. (J) Comparison of PS levels among three states (resting, stimulation, and inhibition) via the GPIb-IX-V signaling pathway. (K) Representative flow cytometric results for CD42b presentation on the platelet surface labeled with PE-conjugated CD42b among three states (resting, stimulation, and inhibition). ( performed on flow cytometry BD FACSAria III; all other flow experiments performed on BD FACSCelesta).
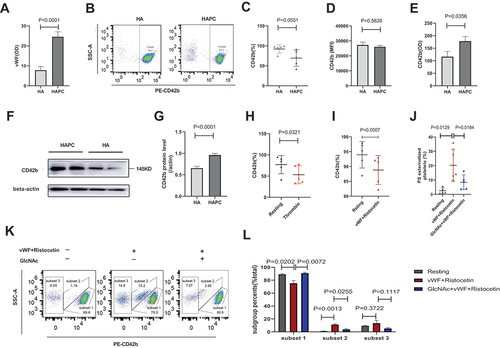
Ristocetin is an antibiotic that promotes binding between vWF and GPIbα [Citation59]. We administered vWF and ristocetin, as well as thrombin, to stimulate purified platelets from the HAPC group, which resulted in a more significant decrease in GPIbα levels on platelet membranes in stimulated platelets compared with resting platelets (88.82 ± 4.85 vs. 93.96 ± 4.48; P = .0007; 53.24 ± 21.29 vs. 77.00 ± 21.59; P = .0321) (). Additionally, we divided the platelets into three subsets after vWF and ristocetin administration. Subset 1 showed higher expression of GPIbα, whereas subset 3 exhibited lower expression. The platelet ratio of subset 2 significantly increased (11.17 ± 2.88 vs. 1.11 ± 0.35; P = .0013) after platelet agonism but decreased (3.83 ± 2.21 vs. 11.17 ± 2.88; P = .0255) after inhibition of GPIbα activation with GlcNAc (). These data revealed that activation of GPIb-IX-V signaling resulted in GPIbα shedding. Furthermore, after administering vWF and ristocetin, PS externalization increased (2.55 ± 2.02 vs. 20.28 ± 11.2; P = .0129), followed by a decrease (20.28 ± 11.2 vs. 8.42 ± 5.16; P = .0184) after GlcNAc treatment to inhibit platelet stimulation (). These findings suggested that vWF and GPIbα interaction was one cause of platelet apoptosis, which was accompanied by GPIbα shedding in the HAPC group. The results of PS exposure in the HAPC group showed certain deviations among three experiments (). One reason for this is that the three experiments were not conducted simultaneously and did not all involve the same participants. Another reason is that the flow cytometers are different. The experiments of used flow cytometer BD FACS Aria, and another two experiments used BD FACS Celesta. Thus, this deviation in results may have resulted from the use of different instruments and parameters.
Discussion
Environmental chronic hypoxia induces erythrocytosis in individuals with intolerance and decreases platelet count. shows that PC was negatively correlated with hemoglobin and hematocrit in patients with HAPC. Previous studies have shown that MKs and erythroid cells in the bone marrow share common progenitor cells [Citation60], while erythrocytosis exposed to long-term hypoxia induces hypomegakaryocytic thrombocytopenia at the expense of expanding the erythroid progenitor cell pool [Citation61]. Moreover, injection of erythropoietin into mice had no effect on PCs, indicating that under chronic exposure to hypoxia, decreases in PCs were not linked to erythrocytosis [Citation62]. The increasing MK count seen here suggests that thrombocytopenia was not induced by excessive differentiation of megakaryocyte-erythroid progenitor into erythroid lineage. Furthermore, MK differentiation, maturation, and apoptosis can be influenced by various oxygen concentrations [Citation63]. This study showed that the maturation of MKs in patients with HAPC was restrained, which was characterized by immature granular MKs instead of mature MKs. What happens to the PC when the number of MKs is increased and the differentiation is restrained? The increased proportion of reticulated platelets in patients with HAPC indicates that the production of platelets is not reduced, while the increased platelet apoptosis is a major cause of the thrombocytopenia in patients with HAPC.
Apoptosis plays an important role in platelet clearance [Citation37]. The levels of PS externalization, MMP, and caspase-3 in platelets shown in correspond to elevated apoptosis in patients with HAPC. Along with the increase in apoptosis, the level of platelet activation was also increased, as shown by elevated platelet P-selectin expression and plasma PF4. Further studies found that the platelet agonists thrombin and vWF were elevated in patients with HAPC, which facilitated platelet activation. After co-incubation with thrombin and vWF, platelet apoptosis increased, suggesting that activation was one of causes of platelet apoptosis in patients with HAPC.
Patients with HAPC exhibit higher erythrocytosis, blood viscosity, and platelet activation, which may increase the incidence of thrombosis. Thrombocytopenia may increase the risk of bleeding when antithrombotic therapy is administered to patients with thrombosis; hence it is important to understand the cause of thrombocytopenia. This study demonstrated that platelet apoptosis is one cause of thrombocytopenia in patients with HAPC. In physiological state, thrombin is a main activator of platelets, and the combination of vWF and GPIbα plays an important role in platelet adhesion and aggregation, both of which can lead to increased platelet apoptosis. For patients with HAPC and thrombosis, is there a key intervention point that can reduce thrombosis and avoid further thrombocytopenia? Indeed, GPIbα could be one such target. In this study, inhibition of GPIba translocation and signaling reduced the increase in platelet apoptosis caused by platelet agonists, as platelet apoptosis is related to platelet membrane surface protein GPIbα, whereas the ligands for the N-terminal ectodomain of platelet GPIbα include thrombin and vWF [Citation64,Citation65]. Thrombin mediates platelet activation primarily by binding to protease-activated receptor-1 on platelets [Citation66,Citation67]. However, subsequent studies showed that GPIbα is necessary for activating platelets via thrombin and proposed that thrombin must bind to GPIbα receptors before protease-activated receptor proteolysis [Citation68–72]. This indicates that GPIbα is required for thrombin-mediated activation of platelets.
These findings provide insight into thrombosis treatment in patients with HAPC and thrombocytopenia. Platelet GPIba is involved in both thrombus formation and platelet apoptosis. Therefore, Anticoagulant therapy targeting GPIbα may be a dual benefit anticoagulant regimen, which is expected to be further verified.
Limitations of the study included using post-chemotherapy remission bone marrow samples as controls to compare the number and classification of MKs in HAPC patients, which could give a false impression of relative reduction in “mature” MK in the HAPC marrows. Despite this, the higher MPV and MK numbers and TO positivity in the HAPC cohort indicate that thrombocytopenia is likely due to peripheral clearance/consumption. Another limitation is the small size of the cohorts in the platelet studies. However, the significance of the effect size partially overpowers this limitation.
Supplemental Material
Download PDF (1.6 MB)Acknowledgments
We thank all members of the Tana Wuren team for their assistance. The authors would also like to thank all members of the Second Department of Gerontology, Qinghai University Affiliated Hospital, for supporting this study, as well as the Physical Examination Department for their cooperation during data collection. We would also like to thank Editage (www.editage.cn) for English language editing.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data supporting the findings of this study are available in the supplementary material.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/09537104.2022.2157381.
Additional information
Funding
References
- Pasha MA, Newman JH. High-altitude disorders: pulmonary hypertension: pulmonary vascular disease: the global perspective. Chest. 2010;137(6 Suppl):13S–11. doi:10.1378/chest.09-2445. PMID: 20522576.
- Leon-Velarde F, Maggiorini M, Reeves JT, Aldashev A, Asmus I, Bernardi L, Ge RL, Hackett P, Kobayashi T, Moore LG, et al. Consensus statement on chronic and subacute high altitude diseases. High Alt Med Biol. 2005;6(2):147–157. doi:10.1089/ham.2005.6.147. PMID: 16060849.
- Bendz B, Rostrup M, Sevre K, Andersen TO, Sandset PM. Association between acute hypobaric hypoxia and activation of coagulation in human beings. Lancet. 2000;356(9242):1657–1658. doi:10.1016/S0140-6736(00)03165-2. PMID: 11089830.
- Cancienne JM, Diduch DR, Werner BC. High altitude is an independent risk factor for postoperative symptomatic venous thromboembolism after knee arthroscopy: a matched case-control study of medicare patients. Arthroscopy. 2017;33(2):422–427. doi:10.1016/j.arthro.2016.07.031. PMID: 27876235.
- Jha PK, Sahu A, Prabhakar A, Tyagi T, Chatterjee T, Arvind P, Nair J, Gupta N, Kumari B, Nair V, et al. Genome-wide expression analysis suggests hypoxia-triggered hyper-coagulation leading to venous thrombosis at high altitude. Thromb Haemost. 2018;118(7):1279–1295. doi:10.1055/s-0038-1657770. PMID: 29864786.
- Ninivaggi M, de Laat M, Lance MM, Kicken CH, Pelkmans L, Bloemen S, Dirks ML, van Loon LJ, Govers-Riemslag JW, Lindhout T, et al. Hypoxia induces a prothrombotic state independently of the physical activity. PLoS One. 2015;10(10):e0141797. doi:10.1371/journal.pone.0141797. PMID: 26516774.
- Tyagi T, Ahmad S, Gupta N, Sahu A, Ahmad Y, Nair V, Chatterjee T, Bajaj N, Sengupta S, Ganju L, et al. Altered expression of platelet proteins and calpain activity mediate hypoxia-induced prothrombotic phenotype. Blood. 2014;123(8):1250–1260. doi:10.1182/blood-2013-05-501924. PMID: 24297866.
- Wang Y, Huang X, Yang W, Zeng Q. Platelets and high-altitude exposure: a meta-analysis. High Alt Med Biol. 2022;23(1):43–56. doi:10.1089/ham.2021.0075. PMID: 35196458.
- Levi M, Lowenberg EC. Thrombocytopenia in critically ill patients. Semin Thromb Hemost. 2008;34(5):417–424. doi:10.1055/s-0028-1092871. PMID: 18956281.
- Levi M, Scully M. How I treat disseminated intravascular coagulation. Blood. 2018;131(8):845–854. doi:10.1182/blood-2017-10-804096. PMID: 29255070.
- Hua VM, Abeynaike L, Glaros E, Campbell H, Pasalic L, Hogg PJ, Chen VM. Necrotic platelets provide a procoagulant surface during thrombosis. Blood. 2015;126(26):2852–2862. doi:10.1182/blood-2015-08-663005. PMID: 26474813.
- Hua VM, Chen VM. Procoagulant platelets and the pathways leading to cell death. Semin Thromb Hemost. 2015;41(4):405–412. doi:10.1055/s-0034-1544002. PMID: 26035696.
- Jackson SP, Schoenwaelder SM. Procoagulant platelets: are they necrotic? Blood. 2010;116(12):2011–2018. doi:10.1182/blood-2010-01-261669. PMID: 20538794.
- Monge CC, Arregui A, Leon-Velarde F. Pathophysiology and epidemiology of chronic mountain sickness. Int J Sports Med. 1992;13:S79–81. doi:10.1055/s-2007-1024603. PMID: 1483802.
- Penaloza D, Arias-Stella J. The heart and pulmonary circulation at high altitudes: healthy highlanders and chronic mountain sickness. Circulation. 2007;115(9):1132–1146. doi:10.1161/CIRCULATIONAHA.106.624544. PMID: 17339571.
- Thiele J, Fischer R. Megakaryocytopoiesis in haematological disorders: diagnostic features of bone marrow biopsies. An overview. Virchows Arch A Pathol Anat Histopathol. 1991;418(2):87–97. doi:10.1007/BF01600283. PMID: 1899960.
- Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, Yost CC, Rubner FJ, Albertine KH, Swoboda KJ, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005;122(3):379–391. doi:10.1016/j.cell.2005.06.015. PMID: 16096058.
- Rondina MT, Schwertz H, Harris ES, Kraemer BF, Campbell RA, Mackman N, Grissom CK, Weyrich AS, Zimmerman GA. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemost. 2011;9(4):748–758. doi:10.1111/j.1538-7836.2011.04208.x. PMID: 21255247.
- Shang C, Wuren T, Ga Q, Bai Z, Guo L, Eustes AS, McComas KN, Rondina MT, Ge R. The human platelet transcriptome and proteome is altered and pro-thrombotic functional responses are increased during prolonged hypoxia exposure at high altitude. Platelets. 2020;31(1):33–42. doi:10.1080/09537104.2019.1572876. PMID: 30721642.
- Handtke S, Thiele T. Large and small platelets—(when) do they differ? J Thromb Haemost. 2020;18(6):1256–1267. doi:10.1111/jth.14788. PMID: 32108994.
- Jackson SR, Carter JM. Platelet volume: laboratory measurement and clinical application. Blood Rev. 1993;7(2):104–113. doi:10.1016/s0268-960x(05)80020-7. PMID: 8369659.
- Kaito K, Otsubo H, Usui N, Yoshida M, Tanno J, Kurihara E, Matsumoto K, Hirata R, Domitsu K, Kobayashi M. Platelet size deviation width, platelet large cell ratio, and mean platelet volume have sufficient sensitivity and specificity in the diagnosis of immune thrombocytopenia. Br J Haematol. 2005;128(5):698–702. doi:10.1111/j.1365-2141.2004.05357.x. PMID: 15725092.
- Ntaios G, Papadopoulos A, Chatzinikolaou A, Saouli Z, Karalazou P, Kaiafa G, Girtovitis F, Kontoninas Z, Savopoulos C, Hatzitolios A, et al. Increased values of mean platelet volume and platelet size deviation width may provide a safe positive diagnosis of idiopathic thrombocytopenic purpura. Acta Haematol. 2008;119(3):173–177. doi:10.1159/000135658. PMID: 18511864.
- Tang YT, He P, Li YZ, Chen HZ, Chang XL, Xie QD, Jiao XY. Diagnostic value of platelet indices and bone marrow megakaryocytic parameters in immune thrombocytopenic purpura. Blood Coagul Fibrinolysis. 2017;28:83–90. doi:10.1097/MBC.0000000000000612. PMID: 35020287.
- Bodrova VV, Shustova ON, Khaspekova SG, Mazurov AV. Platelet reticulated forms, size indexes and functional activity. Interactions in healthy volunteers. Platelets. 2022;33(3):398–403. doi:10.1080/09537104.2021.1922659. PMID: 34029503.
- Hamad MA, Schanze N, Schommer N, Nuhrenberg T, Duerschmied D. Reticulated platelets—Which functions have been established by in vivo and in vitro data? Cells. 2021;10(5):1172. doi:10.3390/cells10051172. PMID: 34065800.
- Hoffmann JJ. Reticulated platelets: analytical aspects and clinical utility. Clin Chem Lab Med. 2014;52(8):1107–1117. doi:10.1515/cclm-2014-0165. PMID: 24807169.
- Bongiovanni D, Santamaria G, Klug M, Santovito D, Felicetta A, Hristov M, von Scheidt M, Aslani M, Cibella J, Weber C, et al. Transcriptome analysis of reticulated platelets reveals a prothrombotic profile. Thromb Haemost. 2019;119(11):1795–1806. doi:10.1055/s-0039-1695009. PMID: 31473989.
- Lim ST, Tobin WO, Murphy S, Kinsella JA, Smith DR, Lim SY, Murphy SM, Coughlan T, Collins DR, O’Neill D, et al. Profile of reticulated platelets in the early, subacute and late phases after transient ischemic attack or ischemic stroke. Platelets. 2022;33(1):89–97. doi:10.1080/09537104.2020.1850670. PMID: 33347340.
- Meershoek AJA, Leunissen TC, van Waes JAR, Klei WA, Huisman A, de Groot MCH, Hoefer IE, van Solinge WW, Moll FL, de Borst GJ. Reticulated platelets as predictor of myocardial injury and 30 day mortality after non-cardiac surgery. Eur J Vasc Endovasc Surg. 2020;59(2):309–318. doi:10.1016/j.ejvs.2019.06.027. PMID: 31812606.
- Wustrow I, Ebner C, Langwieser N, Haller B, Luppa PB, Bradaric C, Bongiovanni D, Stundl A, Laugwitz KL, Ibrahim T, et al. Influence of diagnosis of venous thromboembolism on immature platelets, absolute platelet count and platelet aggregation over time. Platelets. 2021;32(3):398–403. doi:10.1080/09537104.2020.1754380. PMID: 32316806.
- Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1173–1186. doi:10.1016/j.cell.2007.01.037. PMID: 17382885.
- Debrincat MA, Pleines I, Lebois M, Lane RM, Holmes ML, Corbin J, Vandenberg CJ, Alexander WS, Ng AP, Strasser A, et al. BCL-2 is dispensable for thrombopoiesis and platelet survival. Cell Death Dis. 2015;6(4):e1721. doi:10.1038/cddis.2015.97. PMID: 25880088.
- Kodama T, Takehara T, Hikita H, Shimizu S, Shigekawa M, Li W, Miyagi T, Hosui A, Tatsumi T, Ishida H, et al. BH3-only activator proteins Bid and Bim are dispensable for Bak/Bax-dependent thrombocyte apoptosis induced by Bcl-xL deficiency: molecular requisites for the mitochondrial pathway to apoptosis in platelets. J Biol Chem. 2011;286(16):13905–13913. doi:10.1074/jbc.M110.195370. PMID: 2136785.
- Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, Iciek LA, Morgan SJ, Nasarre MC, Nelson R, et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14(5):943–951. doi:10.1038/sj.cdd.4402081. PMID: 17205078.
- Mutlu A, Gyulkhandanyan AV, Freedman J, Leytin V. Concurrent and separate inside-out transition of platelet apoptosis and activation markers to the platelet surface. Br J Haematol. 2013;163(3):377–384. doi:10.1111/bjh.12529. PMID: 24033315.
- Wu LH, Chang SC, Fu TC, Huang CH, Wang JS. High-intensity interval training improves mitochondrial function and suppresses Thrombin generation in platelets undergoing hypoxic stress. Sci Rep. 2017 Jun 23;7(1):4191. doi:10.1038/s41598-017-04035-7. PMID: 28646182.
- Kiers D, Tunjungputri RN, Borkus R, Scheffer GJ, de Groot PG, Urbanus RT, van der Ven AJ, Pickkers P, de Mast Q, Kox M. The influence of hypoxia on platelet function and plasmatic coagulation during systemic inflammation in humans in vivo. Platelets. 2019;30(7):927–930. doi:10.1080/09537104.2018.1557617. PMID: 30584841.
- Rahangdale S, Yeh SY, Novack V, Stevenson K, Barnard MR, Furman MI, Frelinger AL, Michelson AD, Malhotra A. The influence of intermittent hypoxemia on platelet activation in obese patients with obstructive sleep apnea. J Clin Sleep Med. 2011;7:172–178. doi:10.5664/jcsm.28105. PMID: 21509332.
- Leytin V, Allen DJ, Mykhaylov S, Lyubimov E, Freedman J. Thrombin-triggered platelet apoptosis. J Thromb Haemost. 2006;4(12):2656–2663. doi:10.1111/j.1538-7836.2006.02200.x. PMID: 16961585.
- Ferroni P, Martini F, Riondino S, La Farina F, Magnapera A, Ciatti F, Guadagni F. Soluble P-selectin as a marker of in vivo platelet activation. Clin Chim Acta. 2009;399(1–2):88–91. doi:10.1016/j.cca.2008.09.018. PMID: 18835553.
- Theoret JF, Yacoub D, Hachem A, Gillis MA, Merhi Y. P-selectin ligation induces platelet activation and enhances microaggregate and thrombus formation. Thromb Res. 2011;128(3):243–250. doi:10.1016/j.thromres.2011.04.018. PMID: 21600632.
- Quach ME, Chen W, Li R. Mechanisms of platelet clearance and translation to improve platelet storage. Blood. 2018;131(14):1512–1521. doi:10.1182/blood-2017-08-743229. PMID: 29475962.
- Ware J. Molecular analyses of the platelet glycoprotein Ib-IX-V receptor. Thromb Haemost. 1998;79(3):466–478. doi:10.1055/s-0037-1614928. PMID: 9531025.
- Casari C, Du V, Wu YP, Kauskot A, de Groot PG, Christophe OD, Denis CV, de Laat B, Lenting PJ. Accelerated uptake of VWF/platelet complexes in macrophages contributes to VWD type 2b–associated thrombocytopenia. Blood. 2013;122(16):2893–2902. doi:10.1182/blood-2013-03-493312. PMID: 23945153.
- Deng W, Xu Y, Chen W, Paul DS, Syed AK, Dragovich MA, Liang X, Zakas P, Berndt MC, Di Paola J, et al. Platelet clearance via shear-induced unfolding of a membrane mechanoreceptor. Nat Commun. 2016;7(1):12863. doi:10.1038/ncomms12863. PMID: 27670775.
- Gangarosa EJ, Landerman NS, Rosch PJ, Herndon EG, Jr. Hematologic complications arising during ristocetin therapy; relation between dose and toxicity. N Engl J Med. 1958;259(4):156–161. doi:10.1056/NEJM195807242590402. PMID: 13566439.
- Li S, Wang Z, Liao Y, Zhang W, Shi Q, Yan R, Ruan C, Dai K. The glycoprotein Ibα-von Willebrand factor interaction induces platelet apoptosis. J Thromb Haemost. 2010;8(2):341–350. doi:10.1111/j.1538-7836.2009.03653.x. PMID: 19840363.
- Quach ME. Gpib-IX-V and platelet clearance. Platelets. 2021;33(6):1–6. doi:10.1080/09537104.2021.1942815. PMID: 34159884.
- Sanders WE, Read MS, Reddick RL, Garris JB, Brinkhous KM. Thrombotic thrombocytopenia with von Willebrand factor deficiency induced by botrocetin. An animal model. Lab Invest. 1988;59(4):443–452. PMID: 3262788.
- Gitz E, Koopman CD, Giannas A, Koekman CA, van den Heuvel DJ, Deckmyn H, Akkerman JW, Gerritsen HC, Urbanus RT. Platelet interaction with von Willebrand factor is enhanced by shear-induced clustering of glycoprotein Ibalpha. Haematologica. 2013;98(11):1810–1818. doi:10.3324/haematol.2013.087221. PMID: 23753027.
- Kasirer-Friede A, Ware J, Leng L, Marchese P, Ruggeri ZM, Shattil SJ. Lateral clustering of platelet GP Ib-IX complexes leads to up-regulation of the adhesive function of integrin αIIbβ3. J Biol Chem. 2002;277(14):11949–11956. doi:10.1074/jbc.M108727200. PMID: 11812775.
- Shrimpton CN, Borthakur G, Larrucea S, Cruz MA, Dong JF, Lopez JA. Localization of the adhesion receptor glycoprotein Ib-IX-V complex to lipid rafts is required for platelet adhesion and activation. J Exp Med. 2002;196(8):1057–1066. doi:10.1084/jem.20020143. PMID: 12391017.
- Sullam PM, Hyun WC, Szollosi J, Dong J, Foss WM, Lopez JA. Physical proximity and functional interplay of the glycoprotein Ib-IX-V complex and the Fc receptor FcγRIIA on the platelet plasma membrane. J Biol Chem. 1998;273(9):5331–5336. doi:10.1074/jbc.273.9.5331. PMID: 9478992.
- Chen M, Yan R, Zhou K, Li X, Zhang Y, Liu C, Jiang M, Ye H, Meng X, Pang N, et al. Akt-mediated platelet apoptosis and its therapeutic implications in immune thrombocytopenia. Proc Natl Acad Sci USA. 2018;115(45):E10682–10691. doi:10.1073/pnas.1808217115. PMID: 30337485.
- Chen W, Liang X, Syed AK, Jessup P, Church WR, Ware J, Josephson CD, Li R. Inhibiting GPIbalpha shedding preserves post-transfusion recovery and hemostatic function of platelets after prolonged storage. Arterioscler Thromb Vasc Biol. 2016;36:1821–1828. doi:10.1161/ATVBAHA.116.307639. PMID: 27417583.
- Hartley PS, Savill J, Brown SB. The death of human platelets during incubation in citrated plasma involves shedding of CD42b and aggregation of dead platelets. Thromb Haemost. 2006;95:100–106. doi:10.1160/TH05-06-0403. PMID: 16543968.
- Michelson AD, Adelman B, Barnard MR, Carroll E, Handin RI. Platelet storage results in a redistribution of glycoprotein Ib molecules. Evidence for a large intraplatelet pool of glycoprotein Ib. J Clin Invest. 1988;81(6):1734–1740. doi:10.1172/JCI113513. PMID: 3384948.
- Frontroth JP, Favaloro EJ. Ristocetin-induced platelet aggregation (RIPA) and RIPA mixing studies. Methods Mol Biol. 2017;1646:473–494. doi:10.1007/978-1-4939-7196-1_35. PMID: 28804849.
- McDonald TP, Sullivan PS. Megakaryocytic and erythrocytic cell lines share a common precursor cell. Exp Hematol. 1993;21(10):1316–1320. PMID: 8135919.
- Rolovic Z, Basara N, Biljanovic-Paunovic L, Stojanovic N, Suvajdzic N, Pavlovic-Kentera V. Megakaryocytopoiesis in experimentally induced chronic normobaric hypoxia. Exp Hematol. 1990;18(3):190–194. PMID: 2303112.
- Birks JW, Klassen LW, Gurney CW. Hypoxia-induced thrombocytopenia in mice. J Lab Clin Med. 1975;86(2):230–238. PMID: 1151149.
- Mostafa SS, Miller WM, Papoutsakis ET. Oxygen tension influences the differentiation, maturation and apoptosis of human megakaryocytes. Br J Haematol. 2000;111(3):879–889. doi:10.1046/j.1365-2141.2000.02457.x. PMID: 11122151.
- Li R, Emsley J. The organizing principle of the platelet glycoprotein Ib-IX-V complex. J Thromb Haemost. 2013;11(4):605–614. doi:10.1111/jth.12144. PMID: 23336709.
- Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30(12):2341–2349. doi:10.1161/ATVBAHA.110.207522. PMID: 21071698.
- Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103(6):879–887. doi:10.1172/JCI6042. PMID: 10079109.
- Ramachandran R, Noorbakhsh F, Defea K, Hollenberg MD. Targeting proteinase-activated receptors: therapeutic potential and challenges. Nat Rev Drug Discov. 2012;11(1):69–86. doi:10.1038/nrd3615. PMID: 22212680.
- Celikel R, McClintock RA, Roberts JR, Mendolicchio GL, Ware J, Varughese KI, Ruggeri ZM. Modulation of α-Thrombin function by distinct interactions with platelet glycoprotein Ibα. Science. 2003;301(5630):218–221. doi:10.1126/science.1084183. PMID: 12855810.
- De Candia E, Hall SW, Rutella S, Landolfi R, Andrews RK, De Cristofaro R. Binding of thrombin to glycoprotein Ib accelerates the hydrolysis of Par-1 on intact platelets. J Biol Chem. 2001;276(7):4692–4698. doi:10.1074/jbc.M008160200. PMID: 11084032.
- De Cristofaro R, De Candia E, Landolfi R, Rutella S, Hall SW. Structural and functional mapping of the thrombin domain involved in the binding to the platelet glycoprotein Ib. Biochemistry. 2001;40:13268–13273. doi:10.1021/bi010491f. PMID: 11683636.
- Dumas JJ, Kumar R, Seehra J, Somers WS, Mosyak L. Crystal structure of the GpIbα-Thrombin complex essential for platelet aggregation. Science. 2003;301(5630):222–226. doi:10.1126/science.1083917. PMID: 12855811.
- Zarpellon A, Celikel R, Roberts JR, McClintock RA, Mendolicchio GL, Moore KL, Jing H, Varughese KI, Ruggeri ZM. Binding of α-thrombin to surface-anchored platelet glycoprotein Ibα sulfotyrosines through a two-site mechanism involving exosite I. Proc Natl Acad Sci USA. 2011;108(21):8628–8633. doi:10.1073/pnas.1017042108. PMID: 21555542.