
Abstract
CD36 is a multifunctional receptor expressed on the surface of many cell types. Among healthy individuals, CD36 may be absent on platelets and monocytes (type I deficiency) or platelets alone (type II deficiency). However, the exact molecular mechanisms underlying CD36 deficiency remain unclear. In this study, we aimed to identify individuals with CD36 deficiency and investigate the molecular basis underlying it. Blood samples were collected from platelet donors at Kunming Blood Center. Platelets and monocytes were isolated and CD36-expression levels were analyzed using flow cytometry. DNA from whole blood and mRNA isolated from monocytes and platelets of individuals with CD36 deficiency were analyzed using polymerase chain reaction (PCR) testing. The PCR products were cloned and sequenced. Among the 418 blood donors,7 (1.68%) were CD36 deficient: 1 (0.24%) with type I deficiency and 6(1.44%) with type II deficiency. Six heterozygous mutations occurred, including c.268C>T (in type I individuals), c.120 + 1 G>T, c.268C>T, c.329_330del/AC, c.1156 C>T, c.1163A>C, and c.1228_1239del/ATTGTGCCTATT (in type II individuals). Mutations were not detected in one type II individual . At the cDNA level, only mutant, but not wild-type, transcripts were detected in the platelets and monocytes of type I individual. In type II individuals, only mutant transcripts were found in platelets, whereas monocytes possessed wild-type and mutant transcripts. Interestingly, only alternative splicing transcripts were observed in the individual without mutation. We report the incidence rates of type I and II CD36 deficiencies among platelet donors in Kunming. Molecular genetic analyses of DNA and cDNA demonstrated that homozygous mutations on the cDNA level in platelets and monocytes or platelets alone identified type I and II deficiencies, respectively. Furthermore, alternatively spliced products also potentially contribute to the mechanism of CD36 deficiency.
Plain Language Summary
What is the context?
Healthy individuals may lack CD36 on platelets and (or) monocytes, which are defined as Type I and Type II CD36 deficiency. These individuals could develop anti-CD36 antibodies associated with immune-mediated disorders. However, the mechanism underlying the CD36 deficiency is still unclear. In this study, we reported the incidence of CD36 deficiency in Kunming platelet donors and found the new molecular basis of CD36 deficiency individuals.
What’s new?
Molecular genetic analysis of cDNA derived from type I subjects showed the presence of mutant transcript only, both in platelets and monocytes. In type II subjects, platelets only carry mutant transcript, whereas monocytes possessed both wild-type and mutant transcripts. Furthermore, we found that alternatively spliced product of CD36 transcript could also contribute to the mechanism of CD36 deficiencies.
What’s the impact?
Our finding indicates that analysis of CD36 at cDNA level is mandatory to verify different forms of CD36 deficiencies. This information may help us to understand the development of anti-CD36 antibodies in CD36 deficient individuals
Introduction
The cluster of differentiation 36 (CD36), an 88-kDa transmembrane glycoprotein receptor, is expressed in various cell types, including monocytes, macrophages, platelets, microvascular endothelial cells, adipocytes, epithelial cells in the kidney, and cardiac myocytes [Citation1]. CD36 belongs to the class B scavenger receptor family, including scavenger receptor B1 and lysosomal integral membrane protein 2 [Citation1]. The CD36 gene is on chromosome 7q11.2 and comprises 15 exons. Exons 1, 2, 3, and 15 form the 5”untranslated region (UTR) and 3”UTR of CD36 mRNA, while exons 3–14 constitute the open reading frame of CD36 mRNA [Citation2].
Interestingly, CD36 is absent on the platelet surface of certain healthy individuals [Citation3]. CD36 deficiency can be categorized into two: type I deficiency, characterized by a lack of CD36 on platelets and monocytes, and type II deficiency, characterized by a lack of CD36 on platelets only. CD36 deficiency frequency varies across regions and races. It is extremely rare (0.3%) among Caucasians [Citation4,Citation5] but high among Africans (4.82%) [Citation5,Citation6]. In Asia, CD36 frequency varies among Japanese (3.00%–6.80%) [Citation7,Citation8], Thai (2.14%) [Citation9]. The incidence of CD36 deficiency in China ranged between 0.81%–4.13% [Citation10–14], and the most common mutations were 329_330del/AC and c.1228_1239del/ATTGTGCCTATT [Citation14]. Among different Chinese ethnic groups, the largest ethnic group Han has a lower incidence (1.30%), compared with Man (6.45%), Zhuang (3.69%), Bu Yi (3.05%) minorities [Citation11].
Current data indicate that only type I individuals develop anti-CD36 antibodies after platelet transfusion or during pregnancy [Citation15]. Immune-mediated disorders caused by anti-CD36 antibodies have been reported, including platelet transfusion refractoriness (PTR), posttransfusion purpura, fetal/neonatal alloimmune thrombocytopenia (FNAIT) [Citation16–18], and transfusion-related acute lung injury [Citation19]. Meanwhile, accumulated data indicate that anti-CD36 antibodies represent an etiological factor for PTR and the most common platelet antibodies in FNAIT cases in the Chinese population [Citation20].
In this study, we determined the frequency of CD36 deficiencies in Southern China (Kunming, Yunnan province) and analyzed platelet and monocyte DNA and mRNA to determine the molecular basis underlying type I and II CD36 deficiencies.
Materials and methods
Samples
Apheresis platelets extracted from 418 unrelated healthy blood donors between September 2020 and 15 February 2021, were analyzed in this study. Whole blood (approximately 3 mL) was collected and immediately used or preserved at 4°C for 12 h before use. All donors provided informed consent to participate in this study.
Platelet preparation
Platelet-rich plasma was isolated from ethylenediaminetetraacetic acid (EDTA)-anticoagulated blood using differential centrifugation at 120 g for 15 min. Platelets were pelleted using centrifugation at 3,000 g for 15 min to the bottom of the tube, followed by washing twice with phosphate-buffered saline (PBS) containing 0.2% EDTA (PBS-EDTA). Finally, the platelets were suspended in PBS-EDTA buffer at a concentration of 105 platelets/μL.
Flow cytometry analysis
Aliquots of 15 µL platelet suspension previously prepared were incubated with 5 µL fluorescein isothiocyanate (FITC)-conjugated anti-CD36 antibodies (CB38; BD Pharmingen, San Diego, CA, USA) and 2.5 µL phycoerythrin (PE)-labeled anti-CD41 antibodies (Biolegend, San Diego, CA, USA). Platelet-bound antibodies were detected using flow cytometry (Beckman FC500; Pasadena, CA, USA). A negative reaction was defined when≥95% of the platelets did not react with anti-CD36 antibodies. In CD36-negative platelet samples, monocytes were analyzed for CD36 expression. Monocytes were isolated from peripheral blood mononuclear cells using density gradient media (Ficoll-Paque™; Amersham Biosciences, Scituate, MA, USA) and purified using an EasySep™ Human Monocyte Isolation Kit (STEMCELL Technologies Inc., Vancouver, B.C., Canada). Subsequently, viable monocytes were stained with 7-AAD (7-amino-actionomycin D; BD Pharmingen), PE-conjugated anti-CD14 (M5E2, BD Pharmingen) and FITC-labeled anti-CD36 (CB38; BD Pharmingen) and analyzed using flow cytometry as described above.
DNA and cDNA isolations
Genomic DNA was extracted from peripheral blood using a MagCore® Genomic DNA Whole Blood Kit 102 (Reber, Taipei, Taiwan, China). Total RNA was extracted from the platelets and monocytes using TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA) and reverse transcribed into cDNA using an iScript™ cDNA Synthesis Kit (Bio-Rad Laboratories, Berkeley, CA, USA).
CD36 mutation analysis
Genomic DNA from CD36-deficient samples was subjected to sequencing from exon 1 to 15, including each part of the flanking introns, and amplified with specific primers [Citation21]. The polymerase chain reaction (PCR) conditions comprised denaturation at 94°C for 5 min, followed by 32 cycles of denaturation at 94°C for 30 s, annealing at 60.0°C for 30 s, elongation at 72.0°C for 1 min, and a final extension at 72°C for 3 min. cDNA from platelets and monocytes was amplified using two sets of primers, primer-E1–E5 (c.-285_c.430) (forward primer: ACTGCAGTGTAGGACTTTCCTG; reverse primer: CTGCCACAGCCAGATTGAGA), encompassing exons 1–5, and primer-E5–E15 (c.354_c.1567) (forward primer: CTCTTTCCTGCAGCCCAATG and reverse primer: TGGGATGATGGTGTTTCCCC), encompassing exons 5–15. The amplification cycle comprised denaturation at 94°C for 5 min, followed by 40 cycles of denaturation at 94°C for 30 s, annealing at 58.0°C for 30 s, elongation at 72.0°C for 1 min, and a final extension at 72°C for 3 min. PCR products were purified using the EDTA/ethanol precipitation method and sequenced using forward or reverse primers (ABI 3730 ×L Applied Biosystems, Waltham, MA, USA).
Clonal sequencing
PCR products were separated using 2% gel electrophoresis (Thermo Fisher, Waltham, MA, USA) and subsequently ligated into the pGEM®-T vector as recommended by the manufacturer (Tiangen, Beijing, China), transfected into E.coli competent bacteria. Plasmid was subsequently purified using Mini Plasmid Kit (Tiangen), and sequenced (ABI 3730 ×L Applied Biosystems, Waltham, MA, USA).
Results
Frequency of CD36 deficiencies in our cohort
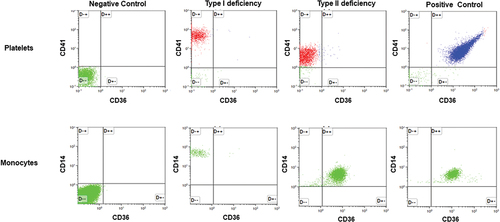
The expression of CD36 on platelets from all donors (n = 418) was measured using flow cytometry (). According to the definition of negative reactions mentioned in the methods section, CD36 expression was undetectable on the platelet surface in seven individuals (two women and five men). Among them, one individual also did not express CD36 on the surface of their monocytes. Therefore, the frequencies of type I and II CD36 deficiencies in our cohort were 0.24%- and 1.44%, respectively.
Figure 1. Flow cytometry analysis of CD36 expression on platelets and monocytes. Top panels: Platelets from a normal individual (positive control); individuals with types I and II deficiencies were stained with fluorescence-labeled anti-CD41 (PE) and anti-CD36 (FITC). After washings, platelets were subjected to flow cytometry analysis using unlabeled platelets as negative controls. Bottom panel: monocytes were isolated from the same individuals and labeled with fluorescence anti-CD14 (PE) and anti-CD36 (FITC) and subjected to flow cytometry analysis as above.
Heterozygous mutations in the DNA of type I and II individuals
Nucleotide sequencing analysis encompassing exons 1–15 and relevant flanking introns of the CD36 gene was performed in all seven individuals with CD36 deficiency. The results are shown in and summarized in . Mutations were detected in six out of seven individuals. A c.268C>T mutation, located on exon 4, was detected in one type I individual (I.1). In five of the six type II individuals (II.1–5), different mutations, including c.120 + 1 G>T (II.1), c.329_330del/AC (II.2), c.1156 C>T (II.3), c.1163A>C (II.4), and c.1228_1239del/ATTGTGCCTATT (II.5), were detected. Interestingly, mutations were present in the heterozygous state, both mutant and wild-type alleles were detected. Mutations were not detected in one type II individual (II.6). Therefore, CD36 absence in these individuals with CD36 deficiency could not be easily explained via DNA analysis.
Figure 2. DNA sequencing analysis of individuals with type I (n = 1) and type II (n = 5) CD36 deficiency. Genomic DNA was isolated and subjected to polymerase chain reaction (PCR) amplification using two primer pairs mentioned in the methods were subsequently analyzed using nucleotide sequencing. The heterozygous mutations (*) detected in the type I individual (I.1) and five type II individuals (II.1–5) are shown. The position of the mutations/deletions are indicated by arrows.
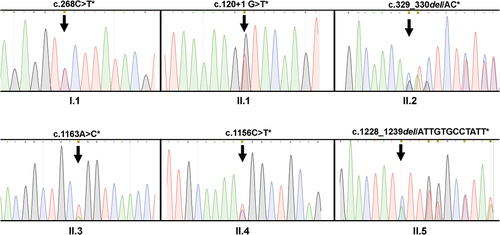
Table I. Summary of the naturally occurring mutations in individuals with CD36 deficiency.
Mutated transcripts were observed in the platelets and monocytes of individuals with type I but only in the platelets of individuals with type II
To clarify the “heterozygous” phenomenon, we compared the transcription patterns underlying CD36 deficiency in platelets and monocytes. mRNA was isolated from platelets and monocytes, reverse-transcribed, and two overlapping regions (exons 1–5 and 5–15) were amplified using RT-PCR. PCR products were separated using gel electrophoresis, and each PCR product was cloned and sequenced. Interestingly, in contrast to the DNA analysis, mutations were detected in the homozygous state in type I individuals. In one type I individual (I.1), only the mutant transcript (c.268T) located on exon 4, but not the wild-type transcript (c.268C), was detected in platelets and monocytes (). Similarly, an analysis of type II individuals (II.2, 4, and 5) revealed that platelets carried only the mutant transcript (homozygous state) (). In contrast, monocytes possess both mutant and wild-type transcripts (heterozygous state). The results are summarized in . Unfortunately, an analysis of the II.3 individual could not be performed owing to limited material.
Figure 3. Sequencing analysis of cDNA from platelets and monocytes derived from individuals with type I (n = 1) and type II (n = 3) CD36 deficiency. Platelet (Plt) and monocyte (Mon) cDNA was amplified using PCR (see Materials and Methods) and subjected to nucleotide sequencing analysis, as described above. Please note that only mutant transcripts were found in the platelets and monocytes of type I individuals. In type II individuals, mutant transcripts were also detected in platelets, whereas monocytes harbored both mutant and wild-type transcripts.
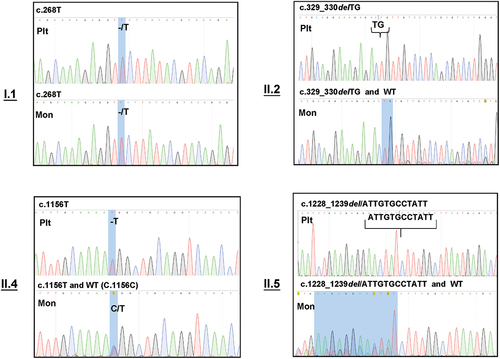
Table II. Summary of the DNA and cDNA sequencing analysis of individuals with CD36 deficiency.
This observation demonstrated that, in contrast to those from type I individuals, monocytes from type II individuals still carried the wild-type CD36 transcript. This phenomenon potentially explains the presence of CD36 on monocyte surfaces.
Alternative splicing product in type II individuals without any detectable mutation
Mutations were not detected in the II.6 individual. Therefore, we analyzed the entire CD36 transcript using PCR amplification of two overlapping regions encompassing exons 1–5 (first region) and 5–15 (second region). When this individual’s platelet and monocyte transcripts were analyzed, no difference was detected in the second region (). As expected, both cell lines expressed normal transcripts (approximately 1200 bp). In contrast, analysis of the first region exhibited a significantly different pattern (). A normal transcript (approximately 750) was observed in monocytes but not in platelets. In contrast, platelets possessed only an alternatively spliced transcript (approximately 400bp) formed by the skipping of exons 2 and 3 (), indicating that monocytes can produce the entire CD36. In contrast, this individual’s platelets could only generate truncated products. Analysis of another individual with type II (II.1) revealed normal transcripts (P0) in monocytes but not in the platelets. In addition, five different alternatively spliced products (P1–P5; around 500, 450, 400, 340, and 250bp) were observed in platelets and monocytes resulting from different exon skipping, including exon 3 (P1); exon 3; c.120_172del in exon 4 (P2); exons 2 and 3 (P3); exons 3 and 4 (P4); and exons 2, 3, and 4 (P5) (). However, in the remaining type II individuals with mutations (II.2, II.4, and II.5), normal transcripts (P0) were observed in the platelets and monocytes (data not shown).
Figure 4. Electrophoresis analysis of PCR products from platelet and monocyte cDNA encompassing exons 1–5 and 5–15. Platelet (Plt) and Monocyte (Mon) cDNA was amplified using PCR (see Materials and Methods) and analyzed with 2% gel electrophoresis using a molecular weight standard as a marker. (A): electrophoresis analysis of exons 1–5, normal transcript (P0), and alternative splice products (P1–P5) are shown; (B) electrophoresis analysis of exons 5–15; the figure shows only one normal band (P0*) in both Plts and Mons.
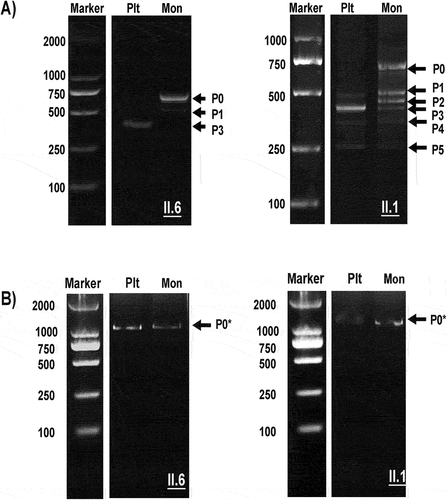
Figure 5. Analysis of alternatively spliced products from type I and II deficiency. PCR products comprising different alternative products (P1–P5) were isolated from the gel, cloned, and sequenced from two directions. The skipping of different exons found in P1, P2, P3, P4, and P5 and mutations are shown.
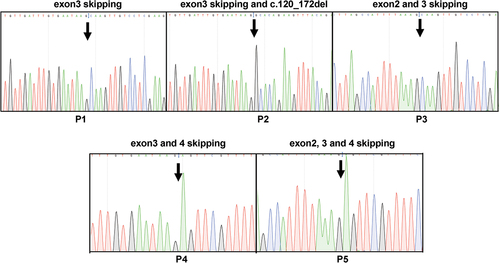
These results indicate that the absence of wild-type transcripts in platelets and monocytes, normal transcripts, and splice variants potentially leads to different types of CD36 deficiency.
Discussion
This study confirmed the lower incidence rate of CD36 deficiency in most provinces located in Southern in China except Guangxi Province (1.68% vs. 4.13%) [Citation11]. In total, we observed six different mutations (c.120 + 1 G>T, c.268C>T, c.329 _330del, c.1156 C>T, c.1163A>C, and c.1228_1239del/ATTGTGCCTATT) associated with CD36 deficiency in six individuals (I.1 and II.1–5) (). To our knowledge, the mutation, c.120 + 1 G>T, found in one type II individual (II.1) has not yet been described. The remaining five mutations have been reported in the Japanese and Chinese populations [Citation4,Citation5,Citation7,Citation8,Citation10,Citation11,Citation18,Citation19].
The type I individual (I.1) was a carrier of the c.268C>T mutation, the most frequent mutation associated with CD36 deficiency in the Japanese population [Citation23]. Interestingly, we found this mutation at the DNA level in the heterozygous state (wild type and mutant allele). However, analysis at the cDNA level revealed that both platelets and monocytes only possessed mutant transcripts (homozygous state). Since wild-type transcript was not found in platelets and monocytes, this finding confirmed the type I deficiency in this individual.
A previous study demonstrated that the c.268C>T mutation resulted into missense mutation Pro90Ser,which affects CD36 maturation, leading to CD36 degradation in the cytoplasm [Citation24]. However, whether this mutation causes complete or only partial degradation of the CD36 protein remains unclear. A recent study in a Japanese population demonstrated that type I individuals carrying the c.268 C>T mutation did not produce anti-CD36 antibodies after platelet transfusion [Citation22]. This observation indicated that platelets (or other CD36-expressing cells) could still express minimal CD36 on the cell surface. Therefore, we presumed that our type I individual carrying c.268C>T mutation also could not produce anti-CD36 antibodies after immunization with wild-type platelets. In this context, whether all type I individuals are at a high risk of developing anti-CD36 antibodies remains intriguing.
The molecular mechanism of type II deficiency is currently less understood than type I. The widely accepted hypothesis is the existence of a “platelet-specific silent allele,” leading to the silencing of CD36 transcription in platelets but not in other cells, such as monocytes [Citation3]. This hypothesis is consistent with our observations. We observed normal transcripts in monocytes but not platelets in individuals with type II deficiency. The monocytes of type II deficient individuals (II.2; II.4 and II.5) carry wild-type and mutant alleles (heterozygous form). We observed only one CD36-positive monocyte population in these individuals. However, the median fluorescence intensity (MFI) of labeled CD36 expressed on these monocytes was significantly lower than in normal individuals (6.54 + 0.03 vs. 16.57 + 3.22; p = .0057). These results indicated allele-dependent CD36 expression on deficient monocytes.Several studies demonstrated that some type II individuals did not harbor any detectable mutations [Citation10,Citation14,Citation25]. The “platelet-specific silent allele” hypothesis could not explain this fact. Similarly, we could not detect any mutation in one of our individuals with type II. In this individual (II.6), we observed normal CD36 transcripts in the monocytes and only alternative splicing products (lacking exons 2 and 3) in the platelets. In addition, we located a mutation “c.120 + 1 G>T” in intron 3 in one individual with type II (II.1), affecting the alternative splicing site and resulting in splicing variants. These truncated products could be degraded during biosynthesis, leading to CD36 absence on the platelet surface, or could not react with anti-CD36 antibodies used in this study. Further study is necessary to verify the effect of this mutation on the expression and structure of CD36. Nevertheless, our observation indicated that alternative splicing mechanisms might also contribute to the emergence of type II deficiency.
Conclusions
In conclusion, we have reported the incidence rates of type I and II CD36 deficiencies in Kunming platelet donors. Molecular genetic analysis demonstrated that homozygous mutations at the cDNA level in platelets and monocytes or platelets alone identified type I and II deficiencies, respectively. Furthermore, we observed that alternatively spliced products might also contribute to the mechanism of CD36 deficiency. However, analysis of a larger cohort applying third-generation sequencing is necessary to validate the mechanism of CD36 deficiency.
Authors’ contributions
QL was responsible for the experimental design and performance, data collection and analysis, and drafting of the original manuscript. MP was responsible for collecting and treating samples from 418 platelet donors at Kunming Blood Center. QC was responsible for the study design. XJ, ZW, QL, ZZ, and ZL assisted with data collection. YY assisted with data analysis. PS, SS, and JW contributed to the study design, data collection, and manuscript revision, and are the corresponding authors.
Ethics approval and consent to participate
The study was approved by the ethics committee of the Institute of Blood Transfusion, Chinese Academy of Medical Sciences.
Acknowledgments
We thank Li Wantong (Panlong District Center for Disease Control and Prevention, Kunming) for her assistance with the data processing. Besides, We would like to thank Editage (www.editage.com) for English language editing.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med. 2014;46(6):e99. doi:10.1038/emm.2014.38.
- Armesilla AL, Vega MA. Structural organization of the gene for human CD36 glycoprotein. J Biol Chem. 1994;269(29):18985–8. doi:10.1016/S0021-9258(17)32263-9.
- Kashiwagi H, Tomiyama Y, Kosugi S, Shiraga M, Lipsky RH, Nagao N, Kanakura Y, Kurata Y, Matsuzawa Y. Family studies of type II CD36 deficient subjects: linkage of a CD36 allele to a platelet-specific mRNA expression defect(s) causing type II CD36 deficiency. Thromb Haemost. 1995;74(02):758–763. doi:10.1055/s-0038-1649809.
- Xia W, Xu X, Fu Y, Ye X, Tsuno N, Santoso S. CD36 deficiency among South-East Asian populations. ISBT Sci Ser. 2016;11(S2):33–36. doi:10.1111/voxs.12261.
- Lee K, Godeau B, Fromont P, Plonquet A, Debili N, Bachir D, Reviron D, Gourin J, Fernandez E, Galacteros F, et al. CD36 deficiency is frequent and can cause platelet immunization in Africans. Transfusion. 1999;39(8):873–879. doi:10.1046/j.1537-2995.1999.39080873.x.
- Curtis BR, Aster RH. Incidence of the Nak(a)-negative platelet phenotype in African Americans is similar to that of Asians. Transfusion. 1996;36(4):331–334. doi:10.1046/j.1537-2995.1996.36496226147.x.
- Ikeda H, Mitani T, Ohnuma M, Haga H, Ohtzuka S, Kato T, Nakase T, Sekiguchi S. A new platelet-specific antigen, Naka, involved in the refractoriness of HLA-matched platelet transfusion. Vox Sang. 1989;57(3):213–217. doi:10.1111/j.1423-0410.1989.tb00826.x.
- Yanai H, Chiba H, Fujiwara H, Morimoto M, Abe K, Yoshida S, Takahashi Y, Fuda H, Hui S-P, Akita H, et al. Phenotype-genotype correlation in CD36 deficiency types I and II. Thromb Haemost. 2000;84(09):436–441. doi:10.1055/s-0037-1614041.
- Phuangtham R, Santoso S, Leelayuwat C, Komvilaisak P, Ding H, Romphruk AV. Frequency of CD36 deficiency in Thais analyzed by quantification of CD36 on cell surfaces and in plasma. Transfusion. 2020;60(4):847–854. doi:10.1111/trf.15737.
- Li R, Qiao Z, Ling B, Lu P, Zhu Z. Incidence and molecular basis of CD36 deficiency in Shanghai population. Transfusion. 2015;55(3):666–673. doi:10.1111/trf.12890.
- Liu J, Shao Y, Ding H, Deng J, Xu X, Wang J, Xia W, Santoso S, Ye X, Fu Y. Distribution of CD36 deficiency in different Chinese ethnic groups. Hum Immunol. 2020;81(7):366–371. doi:10.1016/j.humimm.2020.05.004.
- Lo SC, Lin KH, Hsieh HH, Lin D-T, Hu C-Y. Genetic variations of CD36 and low platelet CD36 expression - a risk factor for lipemic plasma donation in Taiwanese apheresis donors. Vox Sang. 2016;110(3):236–243. doi:10.1111/vox.12356.
- Xu X, Liu Y, Hong X, Chen S, Ma K, Lan X, Ying Y, He J, Zhu F, Lv H. Variants of CD36 gene and their association with CD36 protein expression in platelets. Blood transfusion = Trasfusione del sangue. 2014;12(4):557–564. doi:10.2450/2014.0209-13.
- Xu X, Ye X, Xia W, Liu J, Ding H, Deng J, Chen Y, Shao Y, Wang J, Fu Y, et al. Studies on CD36 deficiency in South China: two cases demonstrating the clinical impact of anti-CD36 antibodies. Thromb Haemost. 2013;110(12):1199–1206. doi:10.1160/TH13-05-0435.
- Lin FQ, Li XF, Li WW, Zhou Z-R, Li J-P. A string of 12 mutations in Intron 3 is a new clue associated with Type II CD36 deficiency. Transfusion. 2019;59(3):1159–1161. doi:10.1111/trf.15084.
- Bierling P, Godeau B, Fromont P, Bettaieb A, Debili N, el-Kassar N, Rouby JJ, Vainchenker W, Duedari N. Posttransfusion purpura-like syndrome associated with CD36 (Naka) isoimmunization. Transfusion. 1995;35(9):777–782. doi:10.1046/j.1537-2995.1995.35996029165.x.
- Fujino H, Ohta K, Taniue J, Nagao N, Hino M, Yamane T, Koh K-R, Takeoka Y, Hirose A, Aoyama Y, et al. Primary refractoriness to platelet transfusion caused by Nak a antibody alone. Vox Sang. 2001;81(1):42–44. doi:10.1046/j.1423-0410.2001.00048.x.
- Xia W, Ye X, Xu X, Ding H, Liu J, Deng J, Chen Y, Shao Y, Wang J, Li H, et al. Two cases of platelet transfusion refractoriness and one case of possible FNAIT caused by antibodies against CD36 in China. Transfus Med. 2014;24(4):254–256. doi:10.1111/tme.12137.
- Nakajima F, Nishimura M, Hashimoto S, Okazaki H, Tadokoro K. Role of anti-Nak a antibody, monocytes and platelets in the development of transfusion-related acute lung injury. Vox Sang. 2008;95(4):318–323. doi:10.1111/j.1423-0410.2008.01095.x.
- Wu G, Zhou Y, Li L, Zhong Z, Li H, Li H, Yu M, Shen W, Ni H. Platelet immunology in China: research and clinical applications. Transfus Med Rev. 2017;31(2):118–125. doi:10.1016/j.tmrv.2016.12.001.
- National Center for Biotechnology Information. PubChem patent summary for CN-108660198-A. [accessed 2019 Sep 30]. https://pubchem.ncbi.nlm.nih.gov/patent/CN-108660198-A.
- Kashiwagi H, Tomiyama Y, Nozaki S, Kiyoi T, Tadokoro S, Matsumoto K, Honda S, Kosugi S, Kurata Y, Matsuzawa Y. Analyses of genetic abnormalities in type I CD36 deficiency in Japan: identification and cell biological characterization of two novel mutations that cause CD36 deficiency in man. Hum Genet. 2001;108(6):459–466. doi:10.1007/s004390100525.
- Masuda Y, Tamura S, Matsuno K, Nagasawa A, Hayasaka K, Shimizu C, Moriyama T. Diverse CD36 expression among Japanese population: defective CD36 mutations cause platelet and monocyte CD36 reductions in not only deficient but also normal phenotype subjects. Thromb Res. 2015;135(5):951–957. doi:10.1016/j.thromres.2015.03.002.
- Kashiwagi H, Tomiyama Y, Honda S, Kosugi S, Shiraga M, Nagao N, Sekiguchi S, Kanayama Y, Kurata Y, Matsuzawa Y. Molecular basis of CD36 deficiency. Evidence that a 478C-->t substitution (proline90-->serine) in CD36 cDNA accounts for CD36 deficiency. J Clin Invest. 1995;95(3):1040–1046. doi:10.1172/JCI117749.
- Hirano K, Kuwasako T, Nakagawa-Toyama Y, Janabi M, Yamashita S, Matsuzawa Y. Pathophysiology of human genetic CD36 deficiency. Trends Cardiovasc Med. 2003;13(4):136–141. doi:10.1016/S1050-1738(03)00026-4.