
Abstract
GALE gene encodes the uridine diphosphate [UDP]-galactose-4-epimerase, which catalyzes the bidirectional interconversion of UDP-glucose to UDP-galactose, and UDP-N-acetyl-glucosamine to UDP-N-acetyl-galactosamine. In that way, GALE balances, through reversible epimerization, the pool of four sugars that are essential during the biosynthesis of glycoproteins and glycolipids. GALE-related disorder presents an autosomal recessive inheritance pattern, and it is commonly associated with galactosemia. Peripheral galactosemia generally associates with non-generalized forms or even asymptomatic presentations, while classical galactosemia may be related to complications such as learning difficulties, developmental delay, cardiac failure, or dysmorphic features. Recently, GALE variants have been related to severe thrombocytopenia, pancytopenia, and in one patient, to myelodysplastic syndrome.
Plain Language Summary
What is the context?
GALE gene encodes for the UDP-Galactose 4-Epimerase, an enzyme involved in the Leloir pathway of galactose catabolism and protein glycosylation.
Homozygous or compound heterozygous GALE variants associate with the disorder known as galactosemia type III.
Three types of galactosemia can be distinguished: the peripheral, the intermediate, and the generalized form, which associate with different clinical symptoms and GALE genetic variants.
Peripheral form is considered benign, while the intermediate and the generalized form is associated with severe and syndromic manifestations, including learning difficulties, delayed growth, sensorineural hearing loss, and early-onset cataracts, among others.
What is new?
In the last few years, GALE variants have been linked to hematological manifestations, such as anemia, febrile neutropenia, and severe thrombocytopenia.
To date, the only GALE variants described in patients presenting hematological disorders are GALE p.Arg51Trp, p.Lys78ValfsX32, p.Val128Met, p.Thr150Met, p.Leu223Pro, and p.Gly237Asp.
The thrombocytopenia observed in GALE patients is associated with reduced GPIbα and β1 integrin glycosylation and externalization to the megakaryocyte and platelet surface, disrupting the actin cytoskeleton remodeling.
What is the impact?
GALE is an essential protein for the correct megakaryocyte and platelet glycosylation.
GALE gene and UDP-galactose 4-epimerase: structure and function
UDP-Galactose 4-Epimerase (GALE) (OMIM: 606953; ENSG00000117308) is a ~ 5 kb gene with 11 exons mapped at chromosome 1p36.11, which encodes for 348 amino acids (aa) protein with a molecular weight of 38 kDa. GALE protein contains a NAD+ binding site, a substrate binding site, and a proton acceptor site ().
Figure 1. Schematic representation of GALE cDNA and protein. (a) Schematic representations of GALE cDNA and UDP-galactose 4-epimerase (GALE protein). GALE encodes a 348 amino acids (aa) protein containing multiple sites for NAD+ binding (blue) (aa from Gly12 to Ile14 (Gly12-Ile14), Asp33-Asn37, Asp66-Ile67, Phe88, Lys92, Lys161, and Tyr185), substrate UDP-glucose binding (green) (aa Ser132-Thr134, Tyr185-Asn187, Asn206-Leu208, Asn224-Phe226, Arg239, and Arg300-Asp304), and a proton acceptor site (yellow) (aa Tyr157). (b) Structural 3D representation of GALE (Rcsb:1ek6). The protein is colored gray, with NAD+ binding sites in light blue, UDP-glucose-binding sites in green, and the proton acceptor site in yellow. NADH molecule and UDP-glucose are represented in dark blue and light green, respectively. Image was created using ChimeraX 1.3 software.
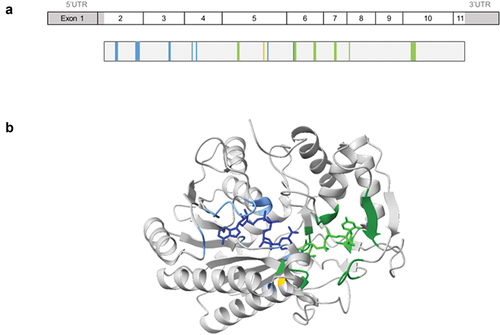
The human enzyme UDP-Galactose 4-Epimerase is a homodimer of two monomers, and a member of the short-chain dehydrogenase/reductase (SDR) family. The catalytic mechanism requires a tightly bound NAD+ cofactor that transiently oxidizes the sugar in the proton acceptor site, and then it is reduced in a non-stereospecific form leading to NADH [Citation1].
This enzyme catalyzes the reversible epimerization of UDP-glucose (Glu) to UDP-galactose (Gal) and the reversible epimerization of UDP-N-acetylglucosamine (GluNAc) to UDP-N-acetylgalactosamine (GalNAc), by binding of any of the four molecules into the substrate binding site. The conversion of UDP-Gal is the final step in the Leloir pathway of galactose catabolism in which the galactose is converted to the glycolytic intermediate glucose 6-phosphate, thus, contributing to the catabolism of dietary galactose [Citation2,Citation3].
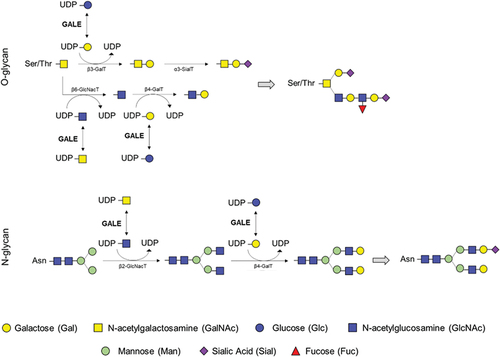
The crucial interconversions of all four of these UDP-hexoses also serve as sugar donors for glycosyltransferase enzymes, so GALE also influences the biosynthesis of glycoproteins and glycolipids in human cells [Citation4]. GALE mediates the protein glycosylation by providing the moieties into the branches during the N- and O-glycosylation in the endoplasmic reticulum (), allowing the formation of mature and functional proteins [Citation5].
Figure 2. Role of the UDP-galactose 4-epimerase in O-linked and N-linked glycosylation. GALE protein allows the interconversion of UDP-glucose and UDP-galactose, and that of UDP-N-acetyl-galactosamine and UDP-N-acetyl-glucosamine. These four molecules are essential in the glycosylation process, by serving as substrates for other enzymes incorporating the carbohydrates of interest and releasing UDP. Branches of carbohydrates are essential in both N-glycosylation and O-glycosylation processes.
Deficiency of UDP-galactose 4-epimerase: clinical and functional phenotype associated with genetic variants
Genetic variants in GALE gene leading to reduced expression or impaired enzyme activity are associated with the inherited metabolic disorder known as epimerase-deficiency galactosemia or galactosemia type III (OMIM number 230350). Three forms of this autosomal recessive disorder have been distinguished, associated with clinical phenotypes that range from nearly benign to life-threatening: the peripheral, the intermediate, and the generalized form.
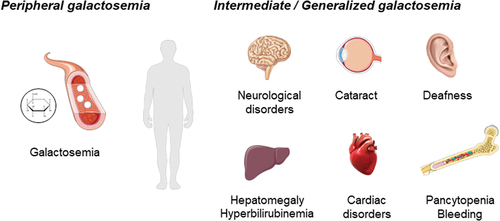
Patients with the peripheral epimerase-deficiency galactosemia exhibit enzyme deficiency that is restricted to the circulating blood cells, and it is considered as clinically benign. It has been described as more frequent in some ethnic groups, such as African-Americans [Citation6]. The common phenotype of these patients is galactosemia, caused by the accumulation of galactose metabolites, and the treatment is mainly based on the restriction of lactose and derivatives. Nevertheless, patients with reduced enzymatic activity without galactosemia had also been described [Citation7]. Otherwise, the generalized form of galactosemia III associates with epimerase deficiency in all tissues, and it is more severe and less common than the peripheral form. Several patients worldwide have been described with syndromic manifestations that may include learning difficulties, delayed growth, sensorineural hearing loss, and early-onset cataracts, and, less frequently, cardiac failure and hepatomegaly [Citation2,Citation8–10] (). Despite the removal of lactose from diet, it is not sufficient to prevent long-term complications [Citation11]. In the last decade, some patients with an intermediate form of galactosemia have been reported some patients with an intermediate form, which present with syndromic manifestations, less marked than in the generalized form, due to an enzyme activity that is markedly deficient in circulating blood cells, but it is higher than 50% compare to normal levels in other cell types [Citation12].
Figure 3. Schematic illustration of the clinical phenotype in patients with peripheral, intermediate, and generalized galactosemia III. in peripheral galactosemia, GALE dysfunction is restricted to the circulating blood cells, especially red blood cells, and the common clinical manifestation is galactosemia. In intermediate/generalized galactosemia, GALE dysfunction can be detected in multiple tissues, and therefore, patients present with syndromic manifestations, that may include neurological disorders, learning difficulties, delay growth, sensorineural hearing loss, early-onset cataracts, cardiac failure, hepatomegaly and hyperbilirubinemia, and pancytopenia associated with increased bleeding tendency. Image resource: Servier Medical Art (https://smart.servier.com/).
To date, around 30 variants in GALE have been described in patients, commonly in compound heterozygosis rather than homozygosis, with different phenotypes (, ).
Figure 4. Variants associated to peripheral, intermediate, and generalized forms of the disease. Reported variants among the GALE protein. Variants marked in red indicate variants causing generalized forms of the disease associated with hematological manifestations. NAD+ binding is represented in blue, substrate UDP-glucose binding in green, and the proton acceptor site in yellow.
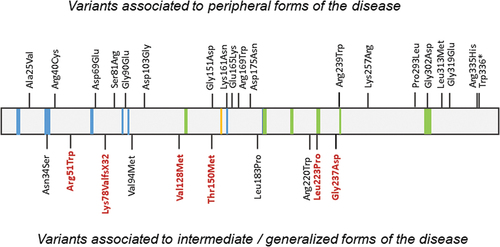
Table I. Molecular, clinical, and laboratory presentation of patients with GALE variants and generalized galactosemia.
The most common variant associated with the severe form of the disease is the p.Val94Met, which was identified in 1999 in a consanguineous family presenting galactosemia, developmental delay, moderate learning difficulties, and hepatomegaly among other clinical symptoms [Citation17]. The same variant was found in a homozygous state in two additional patients with similar clinical features [Citation18]. This variant does not change the overall stability but alters the active site dynamics causing that the substrate, NAD+ cofactor, and binding are less stable [Citation19]. Moreover, Timson DJ characterized several GALE variants in E. Coli, unraveling that the specific activity of the mutant protein p.Val94Met was nearly absent regarding UDP-galactose interconversion, and severely reduced to UDP-N-acetylgalactosamine [Citation13]. This study also demonstrated that variants p.Asn34Ser, p.Gly90Glu, p.Asp103Gly, and p.Leu183Pro were more susceptible to proteolysis than the wild-type protein [Citation13]. However, only p.Asn34Ser and p.Leu183Pro variants have been reported to associate with syndromic manifestations, such as delay in gross motor development and mild-to-moderate mental retardation [Citation14,Citation15] (, ). Of mention, none of the nine variants that have been characterized showed alterations in the enzyme dimerization [Citation13].
Studies in 293T cell lines demonstrated that several GALE variants, such as p.Glu165Lys and p.Trp336*, lead to unstable proteins with reduced half-life and to the presence of aggregates which are partly degraded by the proteasome complex. Moreover, variants p.Arg239Trp and Gly302Asp have no detectable enzyme activity. Authors stated that the altered protein stability is due to its misfolding, and the impaired enzymatic activity is responsible for the molecular defects underlying GALE deficiency [Citation21].
Deficiency of UDP-galactose 4-epimerase and hematological characteristics
Few GALE variants had been associated with hematological alterations. The first evidence of GALE deficiency associated with hematological alterations was reported in 1995 by Rosoff PM, who described a 4-year-old girl with UDP-galactose 4-epimerase deficiency presenting with bruising, thrombocytopenia, and platelet function defect. Moreover, dysplastic cells in the peripheral film and bone marrow changes in the biopsy were observed, so the patient was diagnosed with a myelodysplastic syndrome (MDS). However, no molecular diagnosis was performed and the underlying GALE variants are unknown [Citation22]. The first association of inherited thrombocytopenia with molecular variants in GALE was reported in 2019, in 6 members from a consanguineous family carrying the variant p.Arg51Trp in homozygosis, all of them affected by anemia, febrile neutropenia, and severe thrombocytopenia, associating increased bleeding tendency, without symptoms of systemic galactosemia [Citation16]. One year later, the same variant was identified in compound heterozygosis with p.Gly237Asp in a patient with pancytopenia, cardiac problems, recurrent infections, and splenomegaly [Citation9]. Moreover, in 2021, it was reported a patient with mild macrocytic anemia, leukopenia, thrombocytopenia, mild epistaxis, and increased bilirubin levels, who carried the variant p.Thr150Met in homozygosis [Citation10].
Of mention, none of the patients with pancytopenia and bone marrow dysplasia () have so far developed MDS. However, this patient with p.Thr150Met in homozygosis presented mild megaloblastic changes, which could evolve in the future to SMD [Citation10].
Very recently, we have identified the p.Thr150Met variant in compound heterozygosis with the novel one p.Lys78ValfsX32 in a non-consanguineous family affected with mental retardation, mitral valve prolapse, hyperbilirubinemia, and severe bleeding tendency and macrothrombocytopenia. One of the affected patients had slight leukopenia and mild anemia, while another one presented with erythroblast in blood film since the patient was splenectomized [Citation5]. In addition, we also characterized a second unrelated family with a similar phenotype, carrying the novel variants p.Val128Met and p.Leu223Pro. In this study, we unveiled that these GALE variants are associated with reduced GPIbα and β1 integrin glycosylation and externalization to the megakaryocyte and platelet surface, disrupting the F-actin and filamin A distribution in megakaryocytes, thus affecting the platelet production. Moreover, hypoglycosylated and unfunctional platelets were reported, which were prone to apoptosis, highlighting the key role of GALE in platelet glycosylation, production, and function [Citation5]. Similar to the p.Glu165Lys and p.Trp336* variants described by Bang YL, et al. [Citation21], both p.Val128Met and p.Leu223Pro seem to have an unstable folding leading to reduced protein levels in platelets [Citation5]. Considering that the most GALE variants associated with the generalized forms of the disease are not found in the NAD+ or substrate binding sites (), it is expected that the pathogenicity is due to an abnormal folding and function of the protein.
Overall, loss of GALE activity explains the abnormal glycosylation patterns also seen in some cell cultures and animal models of type III galactosemia [Citation23]. Disease-associated variants have lower activity than the wild-type in red blood cells, and the activity reduction in other tissues is generally greater in variants associated with severe forms of the disease [Citation13,Citation21,Citation24] (). Although the role of GALE in galactose metabolism has been recognized for decades, the biochemical bases of pathophysiology and clinical variability in epimerase deficiency remain unclear, and the nature and severity of the symptoms depend upon a variety of factors that may include environmental factors, such as the dietary restriction of galactose. To date, the mechanisms associated with thrombocytopenia, platelet dysfunction, and bleeding have been unraveled [Citation5], though it is still unknown why some patients with serious syndromic disorders present hematologic disorders, while others do not. Considering the serious syndromic manifestations that patients present, an early diagnosis is essential for the correct treatment, including the withdrawal of dairy milk from the diet, and a periodic review with different medical specialists.
Author contributions
AMQ, JR and JMB wrote the manuscript. All authors critically reviewed the paper.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Chhay JS, Vargas CA, McCorvie TJ, Fridovich-Keil JL, Timson DJ. Analysis of UDP-galactose 4′-epimerase mutations associated with the intermediate form of type III galactosaemia. J Inherit Metab Dis. 2008;31(1):108–8. doi:10.1007/s10545-007-0790-9.
- Demirbas D, Coelho AI, Rubio-Gozalbo ME, Berry GT. Hereditary galactosemia. Metabolism. 2018;83(2018):188–196. doi:10.1016/j.metabol.2018.01.025.
- Fridovich-Keil J, Bean L, He M, et al. Epimerase deficiency galactosemia. In: Adam MP, Everman DB, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2011 Jan 25 [Updated 2021 Mar 4]. p. 1993–2023.
- Broussard A, Florwick A, Desbiens C, Nischan N, Robertson C, Guan Z, Kohler JJ, Wells L, Boyce M. Human UDP-galactose 4′-epimerase (GALE) is required for cell-surface glycome structure and function. J Biol Chem. 2020;295(5):1225–1239. doi:10.1016/S0021-9258(17)49882-6.
- Marín-Quílez A, Di Buduo CA, Díaz-Ajenjo L, Abbonante V, Vuelta E, Soprano PM, Miguel-García C, Santos-Mínguez S, Serramito-Gómez I, Ruiz-Sala P, Peñarrubia MJ, Pardal E, Hernández-Rivas JM, González-Porras JR, García-Tuñón I, Benito R, Rivera J, Balduini A, Bastida JM. Novel variants in GALE cause syndromic macrothrombocytopenia by disrupting glycosylation and thrombopoiesis. Blood. 2023 Jan 26;141(4):406–421. doi:10.1182/blood.2022016995.
- Alano A, Almashanu S, Maceratesi P, Reichardt J, Panny S, Cowan TM. UDP-galactose-4-epimerase deficiency among African-Americans: evidence for multiple alleles. J Invest Med. 1997;5:191A.
- Gitzelmann R. Deficiency of uridine diphosphate galactose 4-epimerase in blood cells of an apparently healthy infant. Preliminary communication. Helv Paediatr Acta. 1972;27(2):125–130.
- Dias Costa F, Ferdinandusse S, Pinto C, Dias A, Keldermans L, Quelhas D, Matthijs G, Mooijer PA, Diogo L, Jaeken J, et al. Galactose epimerase deficiency: expanding the phenotype. JIMD Rep. 2017;37:19–25.
- Febres-Aldana CA, Pelaez L, Wright MS, Maher OM, Febres-Aldana AJ, Sasaki J, Jayakar P, Jayakar A, Diaz-Barbosa M, Janvier M, et al. A case of udp-galactose 4′-epimerase deficiency associated with dyshematopoiesis and atrioventricular valve malformations: an exceptional clinical phenotype explained by altered n-glycosylation with relative preservation of the leloir pathway. Mol Syndromol. 2020;11(5–6):320–330. doi:10.1159/000511343
- Markovitz R, Owen N, Satter LF, Kirk S, Mahoney DH, Bertuch AA, Scaglia F. Expansion of the clinical phenotype of GALE deficiency. Am J Med Genet Part A. 2021;185(10):3118–3121. doi:10.1002/ajmg.a.62384.
- McCorvie TJ, Wasilenko J, Liu Y, Fridovich-Keil JL, Timson DJ. In vivo and in vitro function of human UDP-galactose 4′-epimerase variants. Biochimie. 2011;93(10):1747–1754. doi:10.1016/j.biochi.2011.06.009.
- Openo KK, Schulz JM, Vargas CA, Orton CS, Epstein MP, Schnur RE, Scaglia F, Berry GT, Gottesman GS, Ficicioglu C, et al. Epimerase-deficiency galactosemia is not a binary condition. Am J Hum Genet. 2006;78(1):89–102. doi:10.1086/498985
- Timson DJ. Functional analysis of disease-causing mutations in human UDP-galactose 4-epimerase. FEBS J. 2005;272(23):6170–6177. doi:10.1111/j.1742-4658.2005.05017.x.
- Alano A, Almashanu S, Chinsky JM, Costeas P, Blitzer MG, Wulfsberg EA, Cowan TM. Molecular characterization of a unique patient with epimerase-deficiency galactosaemia. J Inherit Metab Dis. 1998;21(4):341–350. doi:10.1023/A:1005342306080.
- Quimby BB, Alano A, Almashanu S, DeSandro AM, Cowan TM, Fridovich-Keil JL. Characterization of two mutations associated with epimerase-deficiency galactosemia, by use of a yeast expression system for human UDP-galactose-4- epimerase. Am J Hum Genet. 1997;61(3):590–598. doi:10.1086/515517.
- Seo A, Gulsuner S, Pierce S, Ben-Harosh M, Shalev H, Walsh T, Krasnov T, Dgany O, Doulatov S, Tamary H, et al. Inherited thrombocytopenia associated with mutation of UDP-galactose-4-epimerase (GALE). Hum Mol Genet. 2019;28(1):133–142. doi:10.1093/hmg/ddy334
- Wohlers TM, Christacos NC, Harreman MT, Fridovich-Keil JL. Identification and characterization of a mutation, in the human UDP-galactose-4-epimerase gene, associated with generalized epimerase-deficiency galactosemia. Am J Hum Genet. 1999;64(2):462–470. doi:10.1086/302263.
- Wohlers TM, Fridovich-Keil JL. Studies of the V94M-substituted human UDPgalactose-4-epimerase enzyme associated with generalized epimerase-deficiency galactosaemia. J Inherit Metab Dis. 2000;23(7):713–729. doi:10.1023/A:1005682913784.
- Timson DJ, Lindert S. Comparison of dynamics of wildtype and V94M human UDP-galactose 4-epimerase-A computational perspective on severe epimerase-deficiency galactosemia. Gene. 2013;526(2):318–324.
- Liu Y, Bentler K, Coffee B, Chhay JS, Sarafoglou K, Fridovich-Keil JL. A case study of monozygotic twins apparently homozygous for a novel variant of UDP-Galactose 4′-epimerase (GALE): a complex case of variant GALE. JIMD Rep. 2013;7:89–98.
- Bang YL, Nguyen TTT, Trinh TTB, Kim YJ, Song J, Song YH. Functional analysis of mutations in UDP-galactose-4-epimerase (GALE) associated with galactosemia in Korean patients using mammalian GALE-null cells. FEBS J. 2009;276(7):1952–1961. doi:10.1111/j.1742-4658.2009.06922.x.
- Rosoff PM. Myelodysplasia and deficiency of uridine diphosphate-galactose 4-epimerase. J Pediatr. 1995;127(4):605–608. doi:10.1016/S0022-3476(95)70124-9.
- Kingsley DM, Kozarsky KF, Hobble L, Krieger M. Reversible defects in O-linked glycosylation and LDL receptor expression in a UDP-Gal UDP-GalNAc 4-epimerase deficient mutant. Cell. 1986;44(5):749–759. doi:10.1016/0092-8674(86)90841-X.
- McCorvie TJ, Timson DJ. In silico prediction of the effects of mutations in the human UDP-galactose 4’-epimerase gene: towards a predictive framework for type III galactosemia. Gene. 2013;524(2):95–104. doi:10.1016/j.gene.2013.04.061.