
Abstract
Peripheral circadian clocks control cell proliferation and survival, but little is known about their role and regulation in megakaryocytic cells. N-methyl-D-aspartate receptor (NMDAR) regulates the central clock in the brain. The purpose of this study was to determine whether NMDAR regulates the megakaryocytic cell clock and whether the megakaryocytic clock regulates cell proliferation and cell death. We found that both the Meg-01 megakaryocytic cell line and native murine megakaryocytes expressed circadian clock genes. Megakaryocyte-directed deletion of Grin1 in mice caused significant disruption of the circadian rhythm pathway at the transcriptional level and increased expression of BMAL1 at the protein level. Similarly, both pharmacological (MK-801) and genetic (GRIN–/–) inhibition of NMDAR in Meg-01 cells in vitro resulted in widespread changes in clock gene expression including increased expression of BMAL1, the core clock transcription factor. BMAL1 overexpression reduced Meg-01 cell proliferation and altered the time-dependent expression of the cell cycle regulators MYC and WEE1, whereas BMAL1 knockdown led to increased cell death in Meg-01-GRIN1–/– cells. Our results demonstrate that NMDAR regulates the circadian clock in megakaryocytic cells and that the circadian clock component BMAL1 contributes to the control of Meg-01 cell proliferation and survival.
Plain Language Summary
Why was the study done?
Time of day impacts platelet function and production. Our bodies are informed about external time by the brain, but all other cells including platelet precursors megakaryocytes also have their own clock.
Circadian disruption contributes to disorders such as thrombosis (e.g. stroke and heart attacks) and blood cancers (e.g. leukemia). However, the mechanism of circadian control in megakaryocytes remains poorly elucidated.
N-methyl-D-aspartate receptor (NMDAR) regulates circadian clock in the brain and is expressed in megakaryocytes, thus we hypothesized that NMDAR also regulates circadian clock in megakaryocytic cells.
What did the researchers do and find?
We used Meg-01 cell line, its genetically modified version with deleted NMDAR, and data from murine megakaryocytes to determine the NMDAR impact on the clock in these cells.
We found that megakaryocytic cells had all the machinery required to maintain their own circadian clock. NMDAR deletion disrupted circadian clock in megakaryocytic cells.
Manipulation of circadian clock in Meg-01 cells (through BMAL1 overexpression) impacted proliferation and survival of cells.
What do the results mean?
Megakaryocytic cells have their own circadian clock regulated by NMDAR, and its disruption impacts cell proliferation.
What is the objective influence on the wider field?
It is possible that deregulated function of megakaryocytes that occurs in disease can be corrected through the modulation of NMDAR or other components of the cellular circadian clock, thus further studies to develop and test such strategies in disease models are warranted.
Introduction
In the suprachiasmatic nucleus (SCN) of the hypothalamus, N-methyl-D-aspartate receptor (NMDAR) plays a critical role in the generation of circadian rhythm by mediating light-entrainment of the SCN circadian clock via input from retinal ganglion cells.Citation1–3 The SCN clock, or “central” clock, is responsible for daily rhythms in physiology and behaviors such as hormone release and the sleep/wake cycle. Most peripheral cells, including megakaryocytes,Citation4,Citation5 also contain an intrinsic circadian clock termed “peripheral clock” that can be maintained ex vivo independent of the central clock.Citation6 In vivo, the central clock resets peripheral clocks but peripheral clocks are also capable of local cell-autonomous regulation in response to factors such as tissue metabolism or injury.Citation6
Circadian clocks (both central and peripheral) are constructed from a set of autonomous transcription-translation feedback loops (TTFLs).Citation7 The “core clock” is comprised of the positive arm genes (BMAL1, CLOCK, and NPAS2) and the negative arm genes (PER1–3 and CRY1–2). BMAL1 forms a dimer with either CLOCK or NPAS2, and this positive arm complex acts to upregulate the expression of the negative arm genes.Citation8 PER then forms a dimer with CRY, and the negative arm complex acts to inhibit expression of the positive arm genes.Citation8 The core clock is also controlled by auxiliary TTFLs, termed the “auxiliary clock,” as well as other clock gene regulators (such as TIM and CSNK1D) that act either directly or indirectly to modulate core clock protein stabilization or degradation.Citation9,Citation10
Peripheral clocks have an integral role in the control of several cell activities, including cell proliferationCitation3 and the cell cycle.Citation11,Citation12 Compared to the SCN, much less is known about the mechanisms controlling peripheral clocks. However, we recently found that NMDAR regulates the circadian clock in chondrocytes,Citation13,Citation14 suggesting that NMDAR may be involved in regulating both central and peripheral clocks.
NMDARs are glutamate and glycine gated nonspecific cation channels with high permeability to calcium ions (Ca2+).Citation15 NMDARs are hetero-tetrameric structures typically composed of two obligate GluN1 subunits (encoded by the GRIN1 gene) and two modulatory subunits of the GluN2A-D or GluN3A-B types (encoded by GRIN2A-B or GRIN3A-B genes respectively). Multiple studies have shown that NMDAR supports differentiation of normal megakaryocytes,Citation16–19 but proliferation of megakaryocytic leukemia cell lines.Citation20–23 However, the mechanisms involved in these NMDAR effects are not fully understood. Here, we hypothesized that modulation of the peripheral clock contributes to the NMDAR-mediated effects on proliferation and survival of megakaryocytic cells. Elucidation of such mechanisms is important, as findings may help uncover new pathways involved in megakaryocytic disease and suggest novel therapeutic interventions.Citation24
In our previous work, we modeled NMDAR hypo-function in megakaryocytic cells by deleting the GRIN1 gene in Meg-01 cells using the CRISPR-Cas9 systemCitation23 and in mice using the Pf4 promoter.Citation17 In this study, we utilized both the CRISPR-Cas9 modified Meg-01-GRIN1–/– cells and data from Pf4-Grin1–/– mice to confirm the presence of the circadian clock componentry in megakaryocytic cells, determine whether NMDAR regulates the Meg-01 circadian clock, and whether these effects contribute to the mechanisms by which NMDAR affects Meg-01 cell proliferation and survival.
Materials and methods
Cell culture
Meg-01 cellsCitation25 (German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) and Meg-01-GRIN1–/– cellsCitation23 were used as models to study the role of NMDAR in the regulation of intrinsic clock in megakaryocytic cells. Cells were maintained at 37°C, 5% CO2, in RPMI-1640 medium supplemented with 2 mM L-glutamine and 10% fetal bovine serum (FBS; all from Thermo Fisher Scientific, Waltham, Massachusetts, United States). Cells were passaged every 3–4 days; TrypLE (Thermo Fisher Scientific) was used to collect adherent cells. Cells were used up to passage 20. Cultures were confirmed to be free from mycoplasma infection using LookOut Mycoplasma PCR Detection Kit (Sigma–Aldrich).
Transient transfection of plasmids and short interfering RNA
Cells were plated at 2 × 104 cells per well in a 96-well plate and allowed to adhere for 5 h. All transfections were performed in serum-free OptiMEM media (Thermo Fisher Scientific) using lipofectamine transfection reagents following the manufacturer’s protocol. Plasmids (100 ng/well) encoding BMAL1 (Human ARNTL-GFP tagged plasmid RG207870, Origene) or GFP control (a gift from A/Prof James, University of Auckland) were transfected into Meg-01 cells using Lipofectamine 3000 (0.15 μL/well) and P3000 reagent (0.2 μL/well) (Thermo Fisher Scientific). Endoribonuclease-prepared short interfering RNA (esiRNA, 60 nM) targeting BMAL1 (esiARNTL; EHU073641, Sigma-Aldrich) and GFP (esiGFP; EHUEGFP, Sigma-Aldrich) and Lipofectamine RNAimax (0.1 μL/well, Thermo Fisher Scientific) were used to transiently knockdown BMAL1 in Meg-01-GRIN1–/– cells. OptiMEM was replaced with complete culture media 18 h after transfection. The success of transfection was assessed by real-time PCR in cells harvested 48 h later. The percentage of cells successfully transfected was determined by counting GFP-positive cells.
Proliferation and cell death assays
Cell proliferation was assessed as previously described.Citation23 Briefly, the cells were seeded at 1 × 104 cells per well in a 96-well plate. Cells were serum starved for 18 h before re-feeding with serum replete media and culturing for an additional 72 h. Cells were then incubated with bromodeoxyuridine (BrdU) for the final 6 h of the incubation period then assayed with the Cell Proliferation ELISA BrdU kit (Roche, Basel, Switzerland). Cytotoxicity was measured using the Cytotoxicity Detection KitPLUS (lactate dehydrogenase (LDH) release assay; Roche) following the manufacturer’s protocol.
Measurement of relative gene expression in cell lines
Cells were seeded at 1 × 104 cells per well in a 96-well plate. The cells were serum starved for 18 h before re-feeding with serum-replete media. Replicate wells were harvested at 4-h intervals over a 24-h period.
Complementary DNA (cDNA) synthesis and real-time qPCR were performed as previously described.Citation13 cDNA was prepared using a direct cells-to-cDNA protocol. Briefly, cells were lysed with Cell Lysis Buffer II (30 uL/well, Thermo Fisher Scientific) and treated with DNase prior to cDNA synthesis using Moloney Murine Leukemia Virus reverse transcriptase and random primers. Quantitect primer assays were purchased from Qiagen (Hilden, Germany). Primers are detailed in Supplementary Table S1. Real-time qPCR was performed using SYBR Select Master Mix (Thermo Fisher Scientific) on a QuantStudio 5 PCR machine (ThermoFisher Scientific). Relative gene expression was assessed using the 2ΔΔCT method with reference to the housekeeping gene, 18S ribosomal.
Western blotting
Megakaryocytes isolated from the bone marrow of wild-type and Pf4-Grin1–/– mice were lysed in RIPA buffer supplemented with 1 mM each of sodium orthovanadate, sodium fluoride, and PMSF (phenylmethylsulfonyl fluoride). Total protein was measured using the Pierce 660 Assay (Thermo Fisher Scientific) following the manufacturer’s protocol. Equal amounts of protein were loaded on to SDS Page gels and Western blotting undertaken. Blots were probed with anti-BMAL1 primary antibody (catalog number ab2250, Abcam, 1:1000 dilution) for 18 h at 4°C. Following incubation with HRP-conjugated secondary antibodies, proteins were detected by chemiluminescence using a Biorad ChemiDoc Imager and quantified by band densitometry using β-actin as the loading control.
Microarray data analysis
Using the Cre-loxP system, we have recently generated a conditional knockout mouse model of NMDAR function in megakaryocytes.Citation17 The Pf4-promoter-driven Cre recombinase led to the excision of the exons 11 to 22 of the Grin1 gene in megakaryocytes (Pf4-Grin1–/– mice). Mice were housed in an institutional animal facility under controlled conditions (23 ± 1°C, 50 ± 5% humidity, 12-h light/dark cycle) and had free access to food (dry chow) and drinking water. Tissue collection was conducted during the light phase of the light cycle equivalent to zeitgeber time (ZT) 4 to ZT8. Tissue from Pf4-Grin1–/– mice and Pf4-Cre–/– littermate controls (Grin1 wild-type) were processed together. All procedures were approved by the institutional animal ethics committee (approval numbers 001912 and 2516).
Megakaryocytes isolated from the bone marrow of wild-type and Pf4-Grin1–/– mice were subjected to RNA profiling using Clariom S Pico arrays (Thermo Fisher Scientific); results were reported before and deposited in Gene Expression Omnibus repository (GSE183044).Citation17 The current study re-analyzed these data to gain more insights into the expression of circadian clock genes. Hybridization intensity was determined using the Transcriptome Analysis Console 4.0 (Thermo Fisher Scientific). Data were normalized by the gene-level Signal Space Transformation Robust Multi-array (SST-RMA) method, and relative levels of gene expression were determined as before.Citation17,Citation26 Statistical enrichment was determined by ANOVA, with post-hoc analysis using an empirical Bayes model.Citation27 Integration and visualization of data from the Kyoto Encyclopedia of Genes and Genomes (KEGG) circadian rhythm pathway were conducted using the R package Pathview.Citation28
Statistical analysis
Statistical analysis of microarray data is described above. All other experiments were replicated a minimum of three times with data from one experiment constituting one “n” for statistical analysis. Statistical analyses were conducted using GraphPad Prism 8.0 (San Diego, California, United States). Data were tested for normality and homoscedasticity using the Shapiro–Wilk test and Spearman’s test for heteroscedasticity, respectively. For parametric data, differences in group means were compared with either a one-sample Student’s t-test when a single treatment group was being compared to a control group, unpaired t-test when two independent variables were being compared or two-way ANOVA with post-hoc Tukey testing for continuous variables, as indicated in figure legends. Non-parametric data were analyzed using a one-sample Wilcoxon or a Friedman test. For clock gene expression analyses in Meg-01 cells (data shown in ), a more stringent p-value cutoff of <.01 was employed to determine statistical significance to account for multiple genes being measured in the same samples. For all other analyses, a p-value of <.05 was considered statistically significant.
Figure 1. Expression of core clock genes in Meg-01 cells and the effect of NMDAR hypo-function on their expression. Unmodified Meg-01 cells, Meg-01-GRIN1−/− cells, and Meg-01 cells treated with 100 µM MK-801 were serum starved for 18 h prior to re-feeding with serum-replete media. RNA lysates were collected every 4 h for 24 h with lysates for the 0-h time point collected immediately prior to re-feeding. Line graphs show relative expression of (A) positive arm (BMAL1, CLOCK, and NPAS2) and (B) negative arm (PER and CRY families) clock genes over 24 h relative to expression in unmodified Meg-01 cells at 0 h as examined by RT-qPCR. Data represent the mean ± SEM of three biological replicates per group. Statistically significant differences between groups (p < .01) were examined by two-way ANOVA for parametric data and by Friedman’s test for non-parametric data. NMDAR, N-methyl-D-aspartate receptor; RT-qPCR, quantitative reverse transcriptase-polymerase chain reaction.
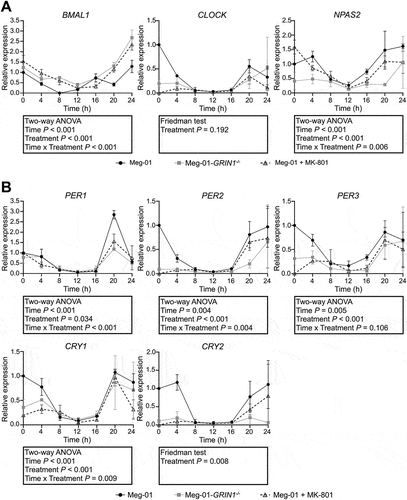
Figure 2. Expression of auxiliary clock genes in Meg-01 cells and the effect of NMDAR hypo-function on their expression. Unmodified Meg-01 cells, Meg-01-GRIN1−/− cells, and Meg-01 cells treated with 100 µM MK-801 were serum starved for 18 h prior to re-feeding with serum-replete media. RNA lysates were collected every 4 h for 24 h; lysates for the 0-h time point were collected immediately prior to re-feeding. Line graphs show expression of (A) auxiliary loop genes (RORA and NR1D1) and (B) core clock gene regulators (TIM and CSNK1D) over 24 h relative to expression in unmodified Meg-01 cells at 0 h as examined by RT-qPCR. Data represent the mean ± SEM of three biological replicates per group. Statistically significant differences between groups (p < .01) were examined by two-way ANOVA for parametric data and by Friedman’s test for non-parametric data. NMDAR, N-methyl-D-aspartate receptor; RT-qPCR, quantitative reverse transcriptase-polymerase chain reaction.
Results
N-methyl-D-aspartate receptor regulates expression of circadian clock genes in Meg-01 cells
First, we sought to determine whether Meg-01 cells express circadian clock components. Meg-01 cells were synchronized by “serum shock.”Citation29 The 0-h timepoint samples were collected immediately prior to refeeding with serum replete media. Meg-01 cells were found to express positive arm genes (BMAL1, CLOCK¸ and NPAS2) and negative arm genes (PER1–3 and CRY1–2) of the core clock (), as well as auxiliary loop clock genes (RORA and NR1D1) and core clock gene regulators (CSNK1D and TIM) (). A statistically significant effect of time was observed for expression of the positive arm core clock genes BMAL1 and NPAS2 (), components of the negative arm PER1, PER2, PER3, and CRY1 (), the auxiliary clock gene NR1D1 () and the core clock gene regulators CSNK1D and TIM () indicating expression of these genes varied over the course of the 24-h period, consistent with the expected time-dependent expression of circadian clock components. The specific effect of time on the expression of CLOCK, CRY2, and RORA could not be assessed as these data were non-parametric ().
Next, we sought to determine whether expression of circadian clock components differed in Meg-01 cells following genetic or pharmacological repression of NMDAR. We found widespread differences in clock gene expression between Meg-01-GRIN1–/– cells and unmodified Meg-01 cells, with statistically significant differences observed for the positive arm core clock genes BMAL1 and NPAS2 (), negative arm core clock genes PER1, PER2, CRY1, and CRY2 () and auxiliary loop genes RORA () and TIM (). However, there was no significant effect of NMDAR deletion on the expression of PER3 (), NR1D1 (), or CSNK1D (). Similar findings were seen in Meg-01 cells treated with the noncompetitive NMDAR antagonist MK-801 (100 μM) ().
Taken together, these data demonstrate that circadian clock genes are expressed in Meg-01 cells in a time-dependent manner, and that NMDAR function contributes to their regulation. Most notable was the effect of NMDAR deletion on the expression of BMAL1, which was higher in both Meg-01-GRIN1–/– cells and MK-801 treated Meg-01 cells across most time points within the 24-h period ().
Overexpression of BMAL1 in Meg-01 cells results in reduced cell proliferation
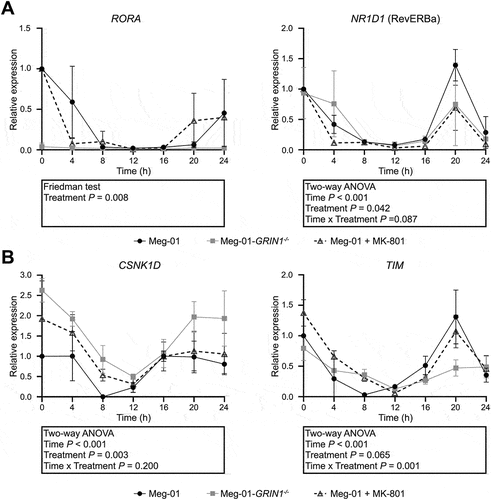
Since BMAL1 showed the most overt difference in expression with NMDAR deletion or MK-801 treatment, we next sought to determine whether elevated BMAL1 expression in these cells contributed to the mechanism by which NMDAR deletion inhibits Meg-01 proliferation. BMAL1 was overexpressed in Meg-01 cells, directionally matching the effect of NMDAR deletion. RT-qPCR analysis after transfection with the BMAL1 overexpression vector confirmed the upregulation of BMAL1 transcripts compared to GFP-transfected controls (p < .001; ). Cell number was significantly lower in BMAL1 overexpressing cells as compared to GFP controls 72-h post transfection (p = .005; ). Concordantly, there was a significant reduction in the BrdU incorporation by BMAL1 overexpressing cells as compared to GFP controls 72-h post transfection (p = .003; ). The mean relative difference in BrdU incorporation between BMAL1 overexpressing cells and GFP controls was 0.74 indicating the number of proliferating cells was approximately 26% lower in BMAL1 overexpressing cells compared to GFP controls. There was no difference in LDH release between BMAL1 overexpressing cells and GFP controls indicating no change in levels of cell death (p = .691; ). Taken together, these data show that BMAL1 overexpression inhibits cell proliferation but does not induce cell death in Meg-01 cells.
Figure 3. Effect of BMAL1 overexpression on Meg-01 cell proliferation and viability. (A) Overexpression of BMAL1 was confirmed by RT-qPCR. Scatter plots showing (B) the relative number of viable cells measured by CyQUANT assay, (C) relative cell proliferation measured by BrdU incorporation assay, and (D) relative cell death measured by LDH release assay. Data points represent individual biological replicates. Statistical significant differences between groups (p < .05) were examined by Student’s t-test. RT-qPCR, quantitative reverse transcriptase-polymerase chain reaction; BrdU, 5-bromo-2-deoxyuridine; LDH, lactate dehydrogenase.
Expression of cell cycle regulators is altered following BMAL1 overexpression in Meg-01 cells
A major mechanism by which the intrinsic clock regulates cell proliferation is via modulating the expression of cell cycle regulators.Citation12 To investigate whether BMAL1 modulated the expression of cell cycle regulators in Meg-01 cells, BMAL1 was again overexpressed in Meg-01 cells. Cells were then synchronized by “serum shock” and the expression of the cell cycle regulator genes MYC, WEE1, and CDKN1A was measured by RT-qPCR in samples collected every 4 h for 24 h.
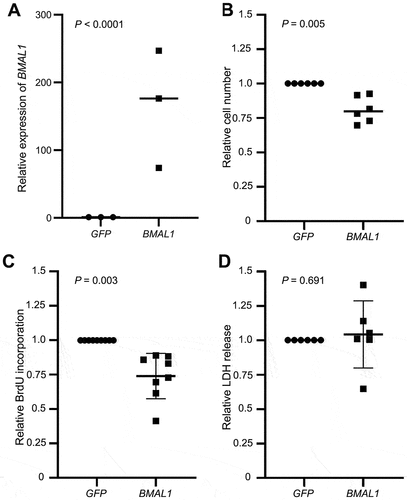
We observed a statistically significant effect of time on the expression of MYC, WEE1, and CDKN1A (p < .001; ) and a significant interaction between time and treatment (BMAL1 overexpression) for MYC and WEE1 (p < .001 for all; ) but not CDKN1A (). These data indicate that BMAL1 regulates MYC and WEE1 expression in Meg-01 cells, but its effects are time-of-day dependent. Levels of both MYC and WEE1, were lower in BMAL1 over-expressing cells at 24 h but not at earlier time points. To exclude the possibility that this effect could have been a result of variation in the level of BMAL1 over-expression over the 24-h period in transfected cells, BMAL1 levels were measured by western blot. We found that levels of BMAL1 were higher in BMAL1-transfected compared to control-transfected cells at every time point across the 24-h period except at 0 h (when cells were still in serum-free media) ().
Figure 4. Effect of BMAL1 overexpression on the expression of cell cycle regulators in Meg-01 cells. Meg-01 cells transfected with a plasmid encoding either BMAL1 or GFP were serum starved for 18 h prior to re-feeding with serum-replete media. RNA lysates were collected every 4 h for 24 h; lysates for the 0-h timepoint were collected immediately prior to re-feeding. Line graphs show relative expression of (A) MYC, (B) WEE1, and (C) CDKN1A over 24 h relative to expression in GFP-transfected controls at 0 h as examined by RT-qPCR. (D) BMAL1 protein levels as measured by western blot in BMAL1-transfected Meg-01 cells compared to GFP transfected controls. Transfected cells were serum starved for 18 h prior to re-feeding with serum-replete media and cell lysates collected every 6 h for 24 h. Lysates for the 0-h time point were collected immediately prior to re-feeding. Representative western blot shown. Quantitative data represent the mean ± SEM of three biological replicates per group. Statistically significant differences between groups (p < .01) were examined by two-way ANOVA. RT-qPCR, quantitative reverse transcriptase-polymerase chain reaction.
BMAL1 knockdown in Meg-01-GRIN1–/–cells does not affect cell proliferation but increases cell death
Since we found that BMAL1 transcript levels were higher in Meg-01-GRIN1–/– cells than in unmodified Meg-01 cells, we next sought to determine the effect of reducing BMAL1 levels in Meg-01-GRIN1–/– cells. To this end, expression of BMAL1 in Meg-01-GRIN1–/– cells was knocked down by transfecting cells with esiRNA targeting BMAL1 (esiBMAL1). Control cells were transfected with esiRNA targeting GFP (esiGFP). Knockdown of BMAL1 was confirmed by RT-qPCR (p < .001; ). We found that cell number was significantly lower in esiBMAL1 transfected Meg-01-GRIN1–/– cells as compared to esiGFP transfected Meg-01-GRIN1–/– cells 72-h post transfection (p = .005; ). There was no significant change in BrdU incorporation by esiBMAL1 transfected Meg-01-GRIN1–/– cells as compared to esiGFP transfected Meg-01-GRIN1–/– cells (p = .72; ); however, esiBMAL1 transfected Meg-01-GRIN1–/– cells had a significantly higher level of LDH release (p = .008; ), suggesting the change in total cell number was due to a change in levels of cell death and not proliferation.
Figure 5. Effect of BMAL1 knockdown on cell proliferation and viability in Meg-01-GRIN1−/− and unmodified Meg-01 cells. (A) knockdown of BMAL1 in Meg-01-GRIN1−/− was confirmed by RT-qPCR. Scatter plots showing (B) the relative number of viable cells measured by CyQUANT assay, (C, E) relative cell proliferation measured by BrdU incorporation assay, and (D, F) relative cell death measured by LDH release assay. Data points represent individual biological replicates. Statistical significant differences between groups (p < .05) were examined by Student’s t-test. RT-qPCR, quantitative reverse transcriptase-polymerase chain reaction; BrdU, 5-bromo-2-deoxyuridine; LDH, lactate dehydrogenase.
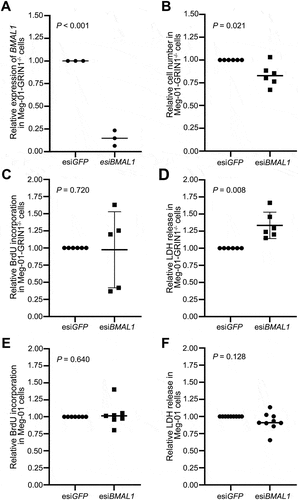
Meg-01-GRIN1–/– cells have elevated levels of ER stress markers and are more susceptible to cell death as compared to unmodified Meg-01 cells.Citation23 Consequently, we next sought to determine whether BMAL1 knockdown also affected cell death in unmodified Meg-01 cells. We found no effect of BMAL1 knockdown on cell proliferation (p = .640; ) or cell death (p = .128; ) in unmodified Meg-01 cells. To determine whether the difference in outcome following BMAL1 knockdown could be due to a difference in BMAL1 levels, we compared BMAL1 expression in Meg-01-GRIN1–/– cells and unmodified Meg-01 cells following transfection with BMAL1-targeting siRNA. We found that BMAL1 RNA levels were not significantly different between Meg-01 and Meg-01-GRIN1–/– cells following BMAL1 knockdown (Suplementary Figure S1).
Grin1 deletion in mouse megakaryocytes alters expression of circadian clock genes
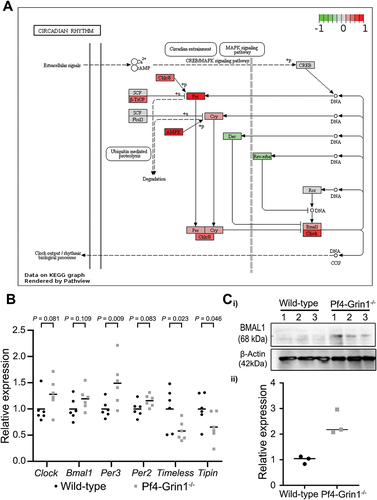
In our previous study, gene expression profiling of CD41-positive megakaryocytes isolated from mouse bone marrow identified that the “Circadian rhythm” pathway (mmu04710) was significantly disrupted in Pf4-Grin1–/– mice compared to wild-type with the normalized enrichment score of 2.56, FDR 0.00, and -Log10(FDR) 2.39.Citation17 In this study, the impact of NMDAR deletion on the circadian rhythm pathway in megakaryocytes was visualized using the Pathview R package () and gene-level changes were investigated further (Supplementary Table S2). A number of circadian clock genes were affected, including the upregulation of Per and Clock, and downregulation of the transcriptional repressors Dec2 and Rev-erbα (Nr1d1). Among the core clock genes, Per3 was the most upregulated (50% higher in Pf4-Grin1–/– megakaryocytes; p = 0.009), and two regulatory genes Timeless (p = .023) and Tipin (p = .046) were the most downregulated (97% and 57% lower in Pf4-Grin1–/– megakaryocytes respectively) (). There was a trend toward upregulation of Clock (46% higher; p = .081), Per2 (17% higher; p = .083), and Bmal1 (21% higher; p = .109). By western blot, protein levels of BMAL1 were found to be significantly higher in Pf4-Grin1–/– megakaryocytes compared to wild type controls (). Taken together, these results highlight significant disruption in the regulation of the circadian clock pathway following NMDAR deletion in megakaryocytes in vivo
Figure 6. Megakaryocyte-directed deletion of Grin1 disrupts clock gene expression in mouse megakaryocytes. (A) scatterplot showing relative transcript levels of select clock genes detected using clariom S pico microarray in Pf4-Grin1–/– megakaryocytes calculated relative to the mean of wild-type megakaryocytes (n = 6 for each). Statistical enrichment was determined by ANOVA with post-hoc analysis using an empirical Bayes model.Citation27 (B) schematics showing transcriptional changes affecting the circadian rhythm pathway (mmu04070) in Pf4-Grin1–/– megakaryocytes relative to wild-type megakaryocytes mapped using the R package pathview. Upregulated molecules are shown in red, downregulated in green, and genes not mapped from our dataset are in gray. Gene abbreviations: AMPK, Prkab1 (protein kinase AMP-activated non-catalytic subunit beta 1), Prkab2 (protein kinase AMP-activated non-catalytic subunit beta 2), Prkag1 (protein kinase AMP-activated non-catalytic subunit gamma 1), Prkag2 (protein kinase AMP-activated non-catalytic subunit gamma 2), Prkag3 (protein kinase AMP-activated non-catalytic subunit gamma 3), Prkaa1 (protein kinase AMP-activated catalytic subunit alpha 1), and Prkaa2 (protein kinase AMP-activated catalytic subunit alpha 2); Bmal1, Arntl (aryl hydrocarbon receptor nuclear translocator-like protein 1); Ck1ε/δ, Csnk1e (casein kinase 1 epsilon), and Csnk1d (casein kinase 1 delta); clock, clock (clock circadian regulator) & Npas2 (neuronal PAS domain protein 2); CREB, Creb1 (cAMP responsive element binding protein 1); Cry, Cry1 (cryptochrome circadian regulator 1) and Cry2 (cryptochrome circadian regulator 2); Dec, Bhlhe40 (basic helix-loop-helix family member E40), and Bhlhe41 (basic helix-loop-helix family member E41); Fbxl3, Fbxl3 (F-box and leucine rich repeat protein 3z); rev-erbα , Nr1d1 (nuclear receptor subfamily 1 group D member 1); Per, Per1 (period circadian regulator 1), Per2 (period circadian regulator 2), and Per3 (period circadian regulator 3); Ror, Rora (RAR-related orphan receptor (a), Rorb (RAR-related orphan receptor (b), Rorc (RAR-related orphan receptor (c); SCF, Skp1a (S-phase kinase-associated protein 1), Cul1 (Cullin 1), and Rbx1 (RING-box protein 1); β-TrCP, Btrc (beta-transducin repeat containing E3 ubiquitin protein ligase), and Fbxw11 (F-box and WD repeat domain containing 11). (C) BMAL1 protein levels (mean ± SEM) as measured by western blotting in Pf4-Grin1–/– megakaryocytes compared to wild type (n = 3). Statistically significant differences in BMAL1 protein levels between WT and Pf4-Grin1–/– megakaryocytes (p < .05) were examined by unpaired t-test.
Discussion
The purpose of this study was to:
demonstrate whether the full panel of circadian clock genes is expressed in the megakaryocytic cell line Meg-01;
establish whether NMDARs regulate clock gene expression in Meg-01 cells and murine megakaryocytes;
determine whether part of the mechanism by which NMDARs regulate Meg-01 cell proliferation may be through regulation of circadian clock components.
We found that Meg-01 cells express all components of the core circadian clock (BMAL1, NPAS2, PER1–3, and CRY1–2), as well as components of the auxiliary clock (RORA and NR1D1), and clock regulators (CSNK1D and TIM) in a time-dependent pattern. Congruent with other studies,Citation4 we also found that bone marrow-derived murine megakaryocytes express circadian clock componentry, demonstrating that both human Meg-01 cells and native murine megakaryocytes contain an intrinsic circadian clock.
Previous studies have shown that mammalian megakaryopoiesis and platelet production display circadian rhythm controlled mostly by the central clock. Human platelet counts peak toward the end of the day (5–7 pm),Citation30–32 whereas platelet activation, aggregation, and certain clotting factors peak in the morning (8–9 am).Citation33–38 In keeping with changes in humans, murine platelet counts peak just after the dark-to-light transition, i.e., at the end of the animals’ active phase equivalent to our end of the day.Citation39 Other parameters showing diurnal/circadian variations in mice include numbers of reticulated platelets, thrombopoietin (TPO) gene (THPO) expression, plasma TPO concentrations, megakaryocyte size, ploidy level, cell cycle activity, and bone marrow TPO receptor (MPL) expression.Citation4,Citation39–42 Global clock gene deletions or mutations disrupt megakaryocyte maturation and platelet function in mice, indicating these processes are under circadian control.Citation41–43 The influence of central versus peripheral clocks over these changes was investigated in mice by comparing the effects of light – versus food – entrainment,Citation4,Citation39 as unlike the central clock, peripheral clocks can be entrained by the timing of food intake.Citation44 It was found that most of the diurnal changes in platelets and megakaryocytes were controlled by the central clock; however, megakaryocyte ploidy and cell cycle activity appeared to be under peripheral clock control,Citation4,Citation39 indicating both central and peripheral clocks are involved in the regulation of megakaryocyte maturation.
NMDAR is known to be a key regulator of the central circadian clockCitation1,Citation2; however, much less is known about how peripheral clocks are controlled. Previously, we showed that NMDAR is involved in the regulation of the circadian clock in chondrocytes, indicating receptor role in regulating peripheral clocks.Citation13,Citation14 In particular, NMDAR blockade disrupted the ability of the chondrocyte circadian clock to respond to serum shock,Citation13,Citation14 an in vitro clock re-setting stimulus.Citation29 This is similar to findings in the present study following serum shock in Meg-01 cells. NMDAR-mediated regulation of the central circadian clock is at least partially related to its role in intracellular calcium homeostasis and the subsequent control of calcium-regulated signaling pathways.Citation45 We have previously shown that similar pathways are involved in regulating the circadian clock in chondrocytes,Citation13,Citation14 suggesting that NMDAR-mediated control of calcium homeostasis may be involved in NMDAR-mediated effects on both central and peripheral clocks.
Using both genetic and pharmacological approaches, this study demonstrates that NMDAR has a widespread effect on the expression of clock genes in both Meg-01 cells and bone marrow-derived murine megakaryocytes. Expression of circadian clock genes was altered in megakaryocytes isolated from Pf4-Grin1–/– mice compared with wild-type mice. However, in contrast to Meg-01-GRIN1–/– cells, most circadian clock genes were upregulated in Pf4-Grin1–/– megakaryocytes. This difference may be influenced by species and cell state differences since mice are nocturnal animals, and leukemic cells can downregulate circadian clock genes.Citation46–48 A consistent finding from both models, however, was that BMAL1 levels were upregulated following GRIN1 deletion, and this was confirmed at the protein level in Pf4-Grin1–/– megakaryocytes. Our previous genome-wide characterization of transcriptomic changes in Meg-01-GRIN1–/– cells and in Pf4-Grin–/– megakaryocytes showed no evidence of a generalized impact of GRIN1 deletion on gene expression to suggest transcriptional effects on clock genes are nonspecific.Citation17,Citation23 Taken together, our data indicate that normal NMDAR function is required to regulate the circadian clock in megakaryocytic cells.
Our previous studies on Meg-01 cells showed that NMDAR deletion (using GRIN1 knockdown) or inhibition (using memantine, MK-801, or AP5) reduced cell proliferation.Citation21–23 Specific NMDAR agonist NMDA increases proliferation of wild-type Meg-01 cells but not of Meg-01-GRIN1–/– cells, validating NMDAR involvement in cell proliferation.Citation23 Here, we found that both NMDAR deletion and inhibition resulted in increased expression of BMAL1. Ectopic overexpression of BMAL1 in Meg-01 cells, to directionally match the observed change in BMAL1 in response to NMDAR hypo-function, led to a reduction in cell proliferation, raising the possibility that part of the mechanism by which NMDAR hypo-function inhibits proliferation is through upregulation of BMAL1.
BMAL1 is a transcription factor and, like other clock components, is known to be involved in controlling the time of day-dependent expression of several target genes, including those encoding cell cycle regulators such as MYC, WEE1, and CDKN1A.Citation3 Here, we found that all three of these genes exhibited time-dependent differences in expression over the course of 24 h, suggesting that these cell cycle regulators may be under circadian control in Meg-01 cells. BMAL1 overexpression led to reduced levels of MYC and WEE1 (but not CDKN1A); however, this effect was only observed at one of the seven time points assessed during the 24-h cycle. This suggests that BMAL1 is involved in controlling the timing of MYC and WEE1 expression in Meg-01 cells. However, BMAL1 effects alone do not fully explain the extent of time-of-day differences in the expression of these genes. As both MYCCitation49,Citation50 and WEE1 are important for driving cell cycle progression, particularly at G2/M,Citation51 reduced expression of either molecule at a single timepoint could potentially impede cell cycle progression and therefore cell proliferation. It is possible that BMAL1 regulation of WEE1 and MYC contributes to the inhibitory effect of BMAL1 overexpression on cell proliferation in Meg-01 cells. However, BMAL1 also has biological activity independent of its ability to regulate transcription. Of particular relevance, BMAL1 physically interacts with the protein translation machinery in the cytosol and reciprocal regulation between BMAL1 and the mTOR signaling pathway is implicated in the circadian control of both cell proliferation and protein translation.Citation52–54 Therefore, BMAL1 may regulate cell proliferation of Meg-01 cells through multiple mechanisms.
Meg-01 cells are derived from a patient with a megakaryocytic subtype of acute myeloid leukemia (AML) transformed from chronic myeloid leukemia (CML) and display a hyper-proliferative phenotype.Citation25 In contrast, Meg-01-GRIN1–/– cells are more differentiated, less proliferative, and show increased levels of cell stress.Citation23 In this study, we found that while overexpression of BMAL1 in Meg-01 cells led to a reduction in cell proliferation, BMAL1 knockdown had no effect on cell proliferation in Meg-01 cells but resulted in increased cell death in Meg-01-GRIN1–/– cells. This difference in outcome may be related to the difference in phenotype of the two cell types. Loss of BMAL1 may have had little effect on cell proliferation in unmodified Meg-01 cells given they already display a hyper-proliferative phenotype, whereas BMAL1 knockdown may have been insufficient to promote cell proliferation in the more differentiated Meg-01-GRIN1–/– cells. Instead, the cell stress phenotype of Meg-01-GRIN1–/– cells may have rendered them vulnerable to the pro-apoptotic effects of BMAL1 knockdown. In support, divergent effects of BMAL1 knockdown on cell proliferation and apoptosis have been reported in a range of other cell types, indicating that the effects of BMAL1 are context/cell-type-dependent.Citation55–61 Of relevance, BMAL1 has been shown to be required for the maintenance of leukemia stem cells in a model of KMT2A:AF9-driven AML, but dispensable for normal hematopoietic stem cell proliferation.Citation62 Thus, the role of BMAL1 in cell proliferation may differ between leukemic and normal progenitors. It remains unknown how BMAL1 levels change during normal megakaryopoiesis and upon leukemic transformation. Previous studies in mice suggest that changes in BMAL1 levels impact megakaryopoiesis. Upon aging, Bmal1-deficient mice develop higher platelet counts accompanied by a higher density of megakaryocytes in the bone marrow.Citation63 Although variable between studies,Citation64 these findings suggest that normal BMAL1 levels curtail megakaryocytic proliferation, which is consistent with our results in Meg-01 cells.
Changes in expression of circadian clock genes occur in CMLCitation46,Citation47 and AML,Citation65,Citation66 and such changes impact pathways involved in leukemia development.Citation67–69 Several previous studies have reported that circadian clock genes, including BMAL1, are downregulated in leukemic cells, which is consistent with their suspected tumor suppressor roles.Citation46–48 To our knowledge, BMAL1 expression has not yet been examined in megakaryocytic malignancies. Our future work will determine BMAL1 levels at different stages of normal megakaryocytic differentiation and in leukemic megakaryoblasts.
Our study contributes novel insights into the regulation and contribution of the intrinsic circadian clock in megakaryocytic cells. Such work is important, as circadian changes occur in several hematologic disorders.Citation24 For example, thrombotic events occur mostly in the morningCitation70,Citation71 and circadian dysregulation impact leukemia cell proliferationCitation69,Citation72 and immune responses.Citation73–77 Megakaryocytes function as immunoregulatory cells,Citation78–80 impacting the course of infections (viral,Citation81,Citation82 bacterial,Citation83 parasiticCitation84), immune-mediated thrombocytopenia,Citation85 bone marrow inflammation, and fibrosis.Citation86,Citation87 Our previous work identified that Pf4-Grin1–/– megakaryocytes display changes in the “Inflammatory mediator regulation of transient receptor potential (TRP) channels” KEGG pathway (mmu04750).Citation17 One could therefore speculate that circadian clock changes may also impact immune roles of megakaryocytes through the mechanism that involves a cross-talk between NMDAR, TRP channels, BMAL1, and inflammatory signaling. This is an interesting area for future investigation, as modulation of cellular circadian machinery offers therapeutic potential. Diverse strategies to target circadian clock components are receiving increasing attention, including against cardiovascular disease,Citation88,Citation89 cancer,Citation90,Citation91 inflammation,Citation92 and infectious diseases.Citation93 NMDAR is amenable to pharmacological modulation, thus NMDAR targeting could be helpful.Citation15 Further studies to dissect the role of circadian clock mechanisms in megakaryocytic cells may advance therapies.
In summary, this study demonstrates that Meg-01 cells contain the full complement of circadian clock componentry, that NMDARs regulate expression of circadian clock components in megakaryocytic cells, and that altered expression of BMAL1 affects Meg-01 cells proliferation and may contribute to the mechanism by which NMDAR deletion causes a reduction in cell proliferation. These data indicate that intrinsic circadian clock regulates the phenotype of Meg-01 cells, raising the possibility that similar mechanisms operate downstream of NMDAR in normal and diseased megakaryocytes. While this study focused on the effects of BMAL1, the expression of multiple other clock genes was also found to be affected following NMDAR deletion and blockade. It is likely that altered expression of these clock components also contributes to the mechanisms by which NMDAR regulates megakaryocytic cell function.
Author contributions
R.C.P. and M.L.K-Z. conceived and designed the study. M.A., J.I.H., M.K., and R.C.P. conducted experimental work. R.C.P, J.I.H., and M.L.K-Z. wrote the paper. All authors approved the final version that has been submitted.
Supplemental Material
Download MS Excel (22.9 KB)Supplemental Material
Download PDF (227.1 KB)Supplemental Material
Download TIFF Image (1.4 MB)Acknowledgments
We are grateful to Nicholas Knowlton for his expert support with microarray data analysis.
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/09537104.2023.2206918.
Additional information
Funding
References
- Ebling FJ. The role of glutamate in the photic regulation of the suprachiasmatic nucleus. Prog Neurobiol. 1996;50(2–3):109–14. doi:10.1016/S0301-0082(96)00032-9.
- Ohi K, Takashima M, Nishikawa T, Takahashi K. N-Methyl-D-Aspartate receptor participates in neuronal transmission of photic information through the retinohypothalamic tract. Neuroendocrinology. 1991;53(4):344–8. doi:10.1159/000125740.
- Brown SA. Circadian clock-mediated control of stem cell division and differentiation: beyond night and day. Development. 2014;141(16):3105–11. doi:10.1242/dev.104851.
- Hartley PS, John Sheward W, French K, Horn JM, Holmes MC, Harmar AJ. Food-entrained rhythmic expression of PER2 and BMAL1 in murine megakaryocytes does not correlate with circadian rhythms in megakaryopoiesis. J Thromb Haemost. 2008;6(7):1144–52. doi:10.1111/j.1538-7836.2008.02978.x.
- Hartley PS. The diurnal tick-tockery of platelet biology. Platelets. 2012;23(2):157–60. doi:10.3109/09537104.2011.600791.
- Richards J, Gumz ML. Advances in understanding the peripheral circadian clocks. FASEB J. 2012;26(9):3602–13. doi:10.1096/fj.12-203554.
- Hastings MH, Maywood ES, Reddy AB. Two decades of circadian time. J Neuroendocrinol. 2008;20(6):812–9. doi:10.1111/j.1365-2826.2008.01715.x.
- Albrecht U. Timing to perfection: the biology of central and peripheral circadian clocks. Neuron. 2012;74(2):246–60. doi:10.1016/j.neuron.2012.04.006.
- Cai YD, Chiu JC. Timeless in animal circadian clocks and beyond. FEBS J. 2022;289(21):6559–75. doi:10.1111/febs.16253.
- Kurien P, Hsu PK, Leon J, Wu D, McMahon T, Shi G, Xu Y, Lipzen A, Pennacchio LA, Jones CR, et al. TIMELESS mutation alters phase responsiveness and causes advanced sleep phase. Proc Natl Acad Sci USA. 2019;116(24):12045–53. doi:10.1073/pnas.1819110116.
- Boucher H, Vanneaux V, Domet T, Parouchev A, Larghero J, Shiels PG. Circadian clock genes modulate human bone marrow mesenchymal stem cell differentiation, migration and cell cycle. PLoS One. 2016;11(1):e0146674. doi:10.1371/journal.pone.0146674.
- Chakrabarti S, Michor F. Circadian clock effects on cellular proliferation: insights from theory and experiments. Curr Opin Cell Biol. 2020;67:17–26. doi:10.1016/j.ceb.2020.07.003.
- Kalev-Zylinska ML, Hearn JI, Rong J, Zhu M, Munro J, Cornish J, Dalbeth N, Poulsen RC. Altered N-methyl D-aspartate receptor subunit expression causes changes to the circadian clock and cell phenotype in osteoarthritic chondrocytes. Osteoarthritis Cartilage. 2018;26(11):1518–30. doi:10.1016/j.joca.2018.06.015.
- Alhilali M, Hearn JI, Rong J, Jain L, Bolam SM, Monk AP, Munro JT, Dalbeth N, Poulsen RC. IL-1beta induces changes in expression of core circadian clock components PER2 and BMAL1 in primary human chondrocytes through the NMDA receptor/CREB and NF-kappaB signalling pathways. Cell Signal. 2021;87:110143. doi:10.1016/j.cellsig.2021.110143.
- Hansen KB, Yi F, Perszyk RE, Furukawa H, Wollmuth LP, Gibb AJ, Traynelis SF. Structure, function, and allosteric modulation of NMDA receptors. J Gen Physiol. 2018;150(8):1081–105. doi:10.1085/jgp.201812032.
- Hitchcock IS, Skerry TM, Howard MR, Genever PG. NMDA receptor-mediated regulation of human megakaryocytopoiesis. Blood. 2003;102(4):1254–9. doi:10.1182/blood-2002-11-3553.
- Hearn JI, Green TN, Hisey CL, Bender M, Josefsson EC, Knowlton N, Baumann J, Poulsen RC, Bohlander SK, Kalev-Zylinska ML. Deletion of Grin1 in mouse megakaryocytes reveals NMDA receptor role in platelet function and proplatelet formation. Blood. 2022;139(17):2673–90. doi:10.1182/blood.2021014000.
- Kalev-Zylinska ML, Hearn JI, Makhro A, Bogdanova A. N-Methyl-D-Aspartate receptors in hematopoietic cells: what have we learned? Front Physiol. 2020;11:577. doi:10.3389/fphys.2020.00577.
- Kalev-Zylinska ML, Morel-Kopp M-C, Ward CM, Hearn JI, Hamilton JR, Bogdanova AY. Ionotropic glutamate receptors in platelets: opposing effects and a unifying hypothesis. Platelets. 2020;32(8):1–11. doi:10.1080/09537104.2020.1852542.
- Genever PG, Wilkinson DJ, Patton AJ, Peet NM, Hong Y, Mathur A, Erusalimsky JD, Skerry TM. Expression of a functional N-methyl-D-aspartate-type glutamate receptor by bone marrow megakaryocytes. Blood. 1999;93(9):2876–83. doi:10.1182/blood.V93.9.2876.409k31_2876_2883.
- Kamal T, Green TN, Hearn JI, Josefsson EC, Morel-Kopp MC, Ward CM, During MJ, Kalev-Zylinska ML. N-methyl-d-aspartate receptor mediated calcium influx supports in vitro differentiation of normal mouse megakaryocytes but proliferation of leukemic cell lines. Res Pract Thromb Haemost. 2018;2(1):125–38. doi:10.1002/rth2.12068.
- Kamal T, Green TN, Morel-Kopp MC, Ward CM, McGregor AL, McGlashan SR, Bohlander SK, Browett PJ, Teague L, During MJ, et al. Inhibition of glutamate regulated calcium entry into leukemic megakaryoblasts reduces cell proliferation and supports differentiation. Cell Signal. 2015;27(9):1860–72. doi:10.1016/j.cellsig.2015.05.004.
- Hearn JI, Green TN, Chopra M, Nursalim YNS, Ladvanszky L, Knowlton N, Blenkiron C, Poulsen RC, Singleton DC, Bohlander SK, et al. N-Methyl-D-Aspartate receptor hypofunction in Meg-01 cells reveals a role for intracellular calcium homeostasis in balancing megakaryocytic-erythroid differentiation. Thromb Haemost. 2020;120(4):671–86. doi:10.1055/s-0040-1708483.
- Pritchett D, Reddy AB. Circadian clocks in the hematologic system. J Biol Rhythms. 2015;30(5):374–88. doi:10.1177/0748730415592729.
- Ogura M, Morishima Y, Ohno R, Kato Y, Hirabayashi N, Nagura H, Saito H. Establishment of a novel human megakaryoblastic leukemia cell line, MEG-01, with positive Philadelphia chromosome. Blood. 1985;66(6):1384–92. doi:10.1182/blood.V66.6.1384.1384.
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):e15. doi:10.1093/nar/gng015.
- Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3(1):Article3. doi:10.2202/1544-6115.1027.
- Luo W, Pant G, Bhavnasi YK, Blanchard SG Jr., Brouwer C. Pathview web: user friendly pathway visualization and data integration. Nucleic Acids Res. 2017;45(W1):W501–w508. doi:10.1093/nar/gkx372.
- Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93(6):929–37. doi:10.1016/S0092-8674(00)81199-X.
- Born J, Lange T, Hansen K, Molle M, Fehm HL. Effects of sleep and circadian rhythm on human circulating immune cells. J Immunol. 1997;158(9):4454–64. doi:10.4049/jimmunol.158.9.4454.
- Kanabrocki EL, Sothern RB, Messmore HL, Roitman-Johnson B, McCormick JB, Dawson S, Bremner FW, Third JL, Nemchausky BA, Shirazi P, et al. Circadian interrelationships among levels of plasma fibrinogen, blood platelets, and serum interleukin-6. Clin Appl Thromb Hemost. 1999;5(1):37–42. doi:10.1177/107602969900500108.
- Bremner WF, Sothern RB, Kanabrocki EL, Ryan M, McCormick JB, Dawson S, Connors ES, Rothschild R, Third JL, Vahed S, et al. Relation between circadian patterns in levels of circulating lipoprotein(a), fibrinogen, platelets, and related lipid variables in men. Am Heart J. 2000;139(1):164–73. doi:10.1016/S0002-8703(00)90324-7.
- Haus E, Cusulos M, Sackett-Lundeen L, Swoyer J. Circadian variations in blood coagulation parameters, alpha-antitrypsin antigen and platelet aggregation and retention in clinically healthy subjects. Chronobiol Int. 1990;7(3):203–16. doi:10.3109/07420529009056976.
- Undar L, Turkay C, Korkmaz L. Circadian variation in circulating platelet aggregates. Ann Med. 1989;21(6):429–33. doi:10.3109/07853898909149234.
- Jafri SM, VanRollins M, Ozawa T, Mammen EF, Goldberg AD, Goldstein S. Circadian variation in platelet function in healthy volunteers. Am J Cardiol. 1992;69(9):951–4. doi:10.1016/0002-9149(92)90799-5.
- Andrews NP, Gralnick HR, Merryman P, Vail M, Quyyumi AA. Mechanisms underlying the morning increase in platelet aggregation: a flow cytometry study. J Am Coll Cardiol. 1996;28(7):1789–95. doi:10.1016/S0735-1097(96)00398-1.
- Tofler GH, Brezinski D, Schafer AI, Czeisler CA, Rutherford JD, Willich SN, Gleason RE, Williams GH, Muller JE. Concurrent morning increase in platelet aggregability and the risk of myocardial infarction and sudden cardiac death. N Engl J Med. 1987;316(24):1514–18. doi:10.1056/NEJM198706113162405.
- Scheer FA, Michelson AD, Frelinger AL, Evoniuk H, Kelly EE, McCarthy M, Doamekpor LA, Barnard MR, Shea SA. The human endogenous circadian system causes greatest platelet activation during the biological morning independent of behaviors. PLoS One. 2011;6(9):e24549. doi:10.1371/journal.pone.0024549.
- Hartley PS, Sheward J, Scholefield E, French K, Horn JM, Holmes MC, Harmar AJ. Timed feeding of mice modulates light-entrained circadian rhythms of reticulated platelet abundance and plasma thrombopoietin and affects gene expression in megakaryocytes. Br J Haematol. 2009;146(2):185–92. doi:10.1111/j.1365-2141.2009.07722.x.
- Hartley PS. Mice housed in groups of 4-6 exhibit a diurnal surge in their platelet count. Platelets. 2013;24(5):412–4. doi:10.3109/09537104.2012.706728.
- Tracey CJ, Pan X, Catterson JH, Harmar AJ, Hussain MM, Hartley PS. Diurnal expression of the thrombopoietin gene is regulated by CLOCK. J Thromb Haemost. 2012;10(4):662–9. doi:10.1111/j.1538-7836.2012.04643.x.
- Zhao Y, Zhang Y, Wang S, Hua Z, Zhang J. The clock gene Per2 is required for normal platelet formation and function. Thromb Res. 2011;127(2):122–30. doi:10.1016/j.thromres.2010.11.025.
- Ohkura N, Oishi K, Sudo T, Hayashi H, Shikata K, Ishida N, Matsuda J, Horie S. CLOCK regulates circadian platelet activity. Thromb Res. 2009;123(3):523–7. doi:10.1016/j.thromres.2008.03.009.
- Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Gen Devel. 2000;14(23):2950–61. doi:10.1101/gad.183500.
- Harvey JRM, Plante AE, Meredith AL. Ion channels controlling circadian rhythms in suprachiasmatic nucleus excitability. Physiol Rev. 2020;100(4):1415–54. doi:10.1152/physrev.00027.2019.
- Yang MY, Chang JG, Lin PM, Tang KP, Chen YH, Lin HY, Liu TC, Hsiao HH, Liu YC, Lin SF. Downregulation of circadian clock genes in chronic myeloid leukemia: alternative methylation pattern of hPER3. Cancer Sci. 2006;97(12):1298–307. doi:10.1111/j.1349-7006.2006.00331.x.
- Yang MY, Yang WC, Lin PM, Hsu JF, Hsiao HH, Liu YC, Tsai HJ, Chang CS, Lin SF. Altered expression of circadian clock genes in human chronic myeloid leukemia. J Biol Rhythms. 2011;26(2):136–48. doi:10.1177/0748730410395527.
- Yang MY, Lin PM, Hsiao HH, Hsu JF, Lin HY, Hsu CM, Chen IY, Su SW, Liu YC, Lin SF. Up-regulation of PER3 expression is correlated with better clinical outcome in acute leukemia. Anticancer Res. 2015;35:6615–22.
- Thompson A, Zhang Y, Kamen D, Jackson CW, Cardiff RD, Ravid K. Deregulated expression of c-myc in megakaryocytes of transgenic mice increases megakaryopoiesis and decreases polyploidization. J Biol Chem. 1996;271(38):22976–82. doi:10.1074/jbc.271.38.22976.
- Guo Y, Niu C, Breslin P, Tang M, Zhang S, Wei W, Kini AR, Paner GP, Alkan S, Morris SW, et al. C-Myc-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Blood. 2009;114(10):2097–106. doi:10.1182/blood-2009-01-197947.
- Kellogg DR. Wee1-dependent mechanisms required for coordination of cell growth and cell division. J Cell Sci. 2003;116(24):4883–90. doi:10.1242/jcs.00908.
- Khapre RV, Kondratova AA, Patel S, Dubrovsky Y, Wrobel M, Antoch MP, Kondratov RV. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging (Albany NY). 2014;6(1):48–57. doi:10.18632/aging.100633.
- Lipton JO, Yuan ED, Boyle LM, Ebrahimi-Fakhari D, Kwiatkowski E, Nathan A, Guttler T, Davis F, Asara JM, Sahin M. The circadian protein BMAL1 regulates translation in response to S6K1-mediated phosphorylation. Cell. 2015;161(5):1138–51. doi:10.1016/j.cell.2015.04.002.
- Lipton JO, Boyle LM, Yuan ED, Hochstrasser KJ, Chifamba FF, Nathan A, Tsai PT, Davis F, Sahin M. Aberrant proteostasis of BMAL1 underlies circadian abnormalities in a paradigmatic Mtor-opathy. Cell Rep. 2017;20(4):868–80. doi:10.1016/j.celrep.2017.07.008.
- Snelling SJ, Forster A, Mukherjee S, Price AJ, Poulsen RC. The chondrocyte-intrinsic circadian clock is disrupted in human osteoarthritis. Chronobiol Int. 2016;33(5):574–9. doi:10.3109/07420528.2016.1158183.
- Yang W, Kang X, Liu J, Li H, Ma Z, Jin X, Qian Z, Xie T, Qin N, Feng D, et al. Clock gene Bmal1 modulates human cartilage gene expression by crosstalk with sirt1. Endocrinology. 2016;157(8):3096–107. doi:10.1210/en.2015-2042.
- Wang J, Li S, Li X, Li B, Li Y, Xia K, Yang Y, Aman S, Wang M, Wu H. Circadian protein BMAL1 promotes breast cancer cell invasion and metastasis by up-regulating matrix metalloproteinase9 expression. Cancer Cell Int. 2019;19(1):182. doi:10.1186/s12935-019-0902-2.
- Korkmaz T, Aygenli F, Emisoglu H, Ozcelik G, Canturk A, Yilmaz S, Ozturk N. Opposite carcinogenic effects of circadian clock gene BMAL1. Sci Rep. 2018;8(1):16023. doi:10.1038/s41598-018-34433-4.
- Zhang Y, Devocelle A, Souza L, Foudi A, Tenreira Bento S, Desterke C, Sherrard R, Ballesta A, Adam R, Giron-Michel J, et al. BMAL1 knockdown triggers different colon carcinoma cell fates by altering the delicate equilibrium between AKT/mTOR and P53/P21 pathways. Aging. 2020;12(9):8067–83. doi:10.18632/aging.103124.
- Ding H, Zhao J, Liu H, Wang J, Lu W. BMAL1 knockdown promoted apoptosis and reduced testosterone secretion in TM3 Leydig cell line. Gene. 2020;747:144672. doi:10.1016/j.gene.2020.144672.
- Sun Y, Wang P, Li H, Dai J. BMAL1 and CLOCK proteins in regulating UVB-induced apoptosis and DNA damage responses in human keratinocytes. J Cell Physiol. 2018;233(12):9563–74. doi:10.1002/jcp.26859.
- Puram RV, Kowalczyk MS, de Boer CG, Schneider RK, Miller PG, McConkey M, Tothova Z, Tejero H, Heckl D, Järås M, et al. Core circadian clock genes regulate leukemia stem cells in AML. Cell. 2016;165(2):303–16. doi:10.1016/j.cell.2016.03.015.
- Hemmeryckx B, Van Hove CE, Fransen P, Emmerechts J, Kauskot A, Bult H, Lijnen HR, Hoylaerts MF. Progression of the prothrombotic state in aging Bmal1-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31(11):2552–9. doi:10.1161/ATVBAHA.111.229062.
- Somanath PR, Podrez EA, Chen J, Ma Y, Marchant K, Antoch M, Byzova TV. Deficiency in core circadian protein Bmal1 is associated with a prothrombotic and vascular phenotype. J Cell Physiol. 2011;226(1):132–40. doi:10.1002/jcp.22314.
- Taniguchi H, Fernandez AF, Setien F, Ropero S, Ballestar E, Villanueva A, Yamamoto H, Imai K, Shinomura Y, Esteller M. Epigenetic inactivation of the circadian clock gene BMAL1 in hematologic malignancies. Cancer Res. 2009;69(21):8447–54. doi:10.1158/0008-5472.CAN-09-0551.
- Rahman S, Al-Hallaj AS, Nedhi A, Gmati G, Ahmed K, Jama HA, Trivilegio T, Mashour A, Askar AA, Boudjelal M. Differential expression of circadian genes in leukemia and a possible role for Sirt1 in restoring the circadian clock in chronic myeloid leukemia. J Circadian Rhythms. 2017;15(1):3. doi:10.5334/jcr.147.
- Gery S, Gombart AF, Yi WS, Koeffler C, Hofmann WK, Koeffler HP. Transcription profiling of C/EBP targets identifies Per2 as a gene implicated in myeloid leukemia. Blood. 2005;106(8):2827–36. doi:10.1182/blood-2005-01-0358.
- Gery S, Koeffler HP. Per2 is a C/EBP target gene implicated in myeloid leukemia. Integr Cancer Ther. 2009;8(4):317–20. doi:10.1177/1534735409352084.
- Jiang H, Yang X, Mi M, Wei X, Wu H, Xin Y, Sun C. PER2: a potential molecular marker for hematological malignancies. Mol Biol Rep. 2021;48(11):7587–95. doi:10.1007/s11033-021-06751-w.
- Marler JR, Price TR, Clark GL, Muller JE, Robertson T, Mohr JP, Hier DB, Wolf PA, Caplan LR, Foulkes MA. Morning increase in onset of ischemic stroke. Stroke. 1989;20(4):473–6. doi:10.1161/01.STR.20.4.473.
- Muller JE, Stone PH, Turi ZG, Rutherford JD, Czeisler CA, Parker C, Poole WK, Passamani E, Roberts R, Robertson T, et al. Circadian variation in the frequency of onset of acute myocardial infarction. N Engl J Med. 1985;313(21):1315–22. doi:10.1056/NEJM198511213132103.
- Sanford ABA, da Cunha LS, Machado CB, de Pinho Pessoa FMC, Silva A, Ribeiro RM, Moreira FC, de Moraes Filho MO, de Moraes MEA, de Souza LEB, et al. Circadian rhythm dysregulation and leukemia development: the role of clock genes as promising biomarkers. Int J Mol Sci. 2022;23(15):8212. doi:10.3390/ijms23158212.
- Ince LM, Barnoud C, Lutes LK, Pick R, Wang C, Sinturel F, Chen CS, de Juan A, Weber J, Holtkamp SJ, et al. Influence of circadian clocks on adaptive immunity and vaccination responses. Nat Commun. 2023;14(1):476. doi:10.1038/s41467-023-35979-2.
- Cervantes-Silva MP, Carroll RG, Wilk MM, Moreira D, Payet CA, O’Siorain JR, Cox SL, Fagan LE, Klavina PA, He Y, et al. The circadian clock influences T cell responses to vaccination by regulating dendritic cell antigen processing. Nat Commun. 2022;13(1):7217. doi:10.1038/s41467-022-34897-z.
- Wang C, Lutes LK, Barnoud C, Scheiermann C. The circadian immune system. Sci Immunol. 2022;7(72):eabm2465. doi:10.1126/sciimmunol.abm2465.
- Hong H, Cheung YM, Cao X, Wu Y, Li C, Tian XY. REV-ERBalpha agonist SR9009 suppresses IL-1beta production in macrophages through BMAL1-dependent inhibition of inflammasome. Biochem Pharmacol. 2021;192:114701. doi:10.1016/j.bcp.2021.114701.
- Deng W, Zhu S, Zeng L, Liu J, Kang R, Yang M, Cao L, Wang H, Billiar TR, Jiang J, et al. The circadian clock controls immune checkpoint pathway in sepsis. Cell Rep. 2018;24(2):366–78. doi:10.1016/j.celrep.2018.06.026.
- Cunin P, Nigrovic PA. Megakaryocytes as immune cells. J Leukoc Biol. 2019;105(6):1111–21. doi:10.1002/JLB.MR0718-261RR.
- Tilburg J, Becker IC, Italiano JE. Don’t you forget about me(gakaryocytes). Blood. 2022;139(22):3245–54. doi:10.1182/blood.2020009302.
- Koupenova M, Livada AC, Morrell CN. Platelet and megakaryocyte roles in innate and adaptive immunity. Circ Res. 2022;130(2):288–308. doi:10.1161/CIRCRESAHA.121.319821.
- Bernardes JP, Mishra N, Tran F, Bahmer T, Best L, Blase JI, Bordoni D, Franzenburg J, Geisen U, Josephs-Spaulding J, et al. Longitudinal multi-omics analyses identify responses of megakaryocytes, erythroid cells, and plasmablasts as hallmarks of severe COVID-19. Immunity. 2020;53(6):1296–314 e1299. doi:10.1016/j.immuni.2020.11.017.
- Campbell RA, Schwertz H, Hottz ED, Rowley JW, Manne BK, Washington AV, Hunter-Mellado R, Tolley ND, Christensen M, Eustes AS, et al. Human megakaryocytes possess intrinsic antiviral immunity through regulated induction of IFITM3. Blood. 2019;133(19):2013–26. doi:10.1182/blood-2018-09-873984.
- Wang J, Xie J, Wang D, Han X, Chen M, Shi G, Jiang L, Zhao M. Cxcr4(high) megakaryocytes regulate host-defense immunity against bacterial pathogens. Elife. 2022;11:11. doi:10.7554/eLife.78662.
- Wunderlich F, Delic D, Gerovska D, Arauzo-Bravo MJ. Vaccination accelerates liver-intrinsic expression of megakaryocyte-related genes in response to blood-stage malaria. Vaccines (Basel). 2022;10(2):287. doi:10.3390/vaccines10020287.
- Liu Y, Zuo X, Chen P, Hu X, Sheng Z, Liu A, Liu Q, Leng S, Zhang X, Li X, et al. Deciphering transcriptome alterations in bone marrow hematopoiesis at single-cell resolution in immune thrombocytopenia. Signal Transduct Target Ther. 2022;7(1):347. doi:10.1038/s41392-022-01167-9.
- Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O’Donnell E, Farndale RW, Ware J, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327(5965):580–3. doi:10.1126/science.1181928.
- Khatib-Massalha E, Mendez-Ferrer S. Megakaryocyte diversity in ontogeny, functions and cell-cell interactions. Front Oncol. 2022;12:840044. doi:10.3389/fonc.2022.840044.
- Oyama Y, Walker LA, Eckle T. Targeting circadian PER2 as therapy in myocardial ischemia and reperfusion injury. Chronobiol Int. 2021;38(9):1262–73. doi:10.1080/07420528.2021.1928160.
- Penaloza-Martinez E, Moreno G, Aroca-Crevillen A, Huertas S, Vicent L, Rosillo N, Hidalgo A, Bueno H. Circadian rhythms in thrombosis and atherothrombotic events. Front Biosci (Landmark Ed). 2022;27(2):51. doi:10.31083/j.fbl2702051.
- Battaglin F, Chan P, Pan Y, Soni S, Qu M, Spiller ER, Castanon S, Roussos Torres ET, Mumenthaler SM, Kay SA, et al. Clocking cancer: the circadian clock as a target in cancer therapy. Oncogene. 2021;40(18):3187–200. doi:10.1038/s41388-021-01778-6.
- Ruan W, Yuan X, Eltzschig HK. Circadian rhythm as a therapeutic target. Nat Rev Drug Discov. 2021;20(4):287–307. doi:10.1038/s41573-020-00109-w.
- Poole J, Ray D. The role of circadian clock genes in critical illness: the potential role of translational clock gene therapies for targeting inflammation, mitochondrial function, and muscle mass in intensive care. J Biol Rhythms. 2022;37(4):385–402. doi:10.1177/07487304221092727.
- Xia Y, Ding X, Wang S, Ren W. Circadian orchestration of host and gut microbiota in infection. Biol Rev Camb Philos Soc. 2023;98(1):115–31. doi:10.1111/brv.12898.