
Abstract
ANKRD26-related thrombocytopenia (ANKRD26-RT or THC2, MIM 188 000), an autosomal dominant thrombocytopenia, is unresponsive to immunosuppressive therapy and susceptible to hematological malignancies. A large number of pediatric patients are diagnosed with immune thrombocytopenia (ITP) every year; however, thrombocytopenia of genetic origin is often missed. Extensive characterization of ANKRD26-RT will help prevent missed diagnosis and misdiagnosis. Furthermore, identification of ANKRD26-RT will help in the formulation of an accurate diagnosis and a treatment plan. In our study, we report cases of two Chinese pediatric patients with ANKRD26-RT and analyze their clinical characteristics, gene mutations, and treatment modalities. Both patients were 1-year-old and presented with mild bleeding (World Health Organization(WHO) score grade 1), different degrees of platelet reduction, normal mean platelet volume, and megakaryocyte maturation impairment not obvious. Genetic tests revealed that both patients had ANKRD26 gene mutations.Patient 1 had a mutation c.-140C>G of the 5’ untranslated region (UTR), and patient 2 had a mutation of c.-127A>T of 5’UTR. Both patients were treated with eltrombopag, and the treatment was no response, with no adverse reactions.
Plain Language Summary
What is the background?
ANKRD26-RT is an autosomal dominant thrombocytopenia which is unresponsive to immunosuppressive therapy and susceptible to hematological malignancies.
It is rare and lacks specific clinical features, making misdiagnosis easy.
Some studies report that eltrombopag is safe and effective for short-term treatment of the disease; however, these reports are limited.
What we did and summary of findings.
We retrospectively studied the clinical manifestations and diagnosis process of ANKRD26-RT and discussed the treatment efficacy of immunosuppressants and eltrombopag for its management.
We found two pediatric cases of patients with ANKRD26-RT with varying degrees of thrombocytopenia, mild bleeding, normal mean platelet volume, and megakaryocyte maturation impairment that was not obvious. Immunosuppressant treatment wasunresponsiveor temporarily responsivebut not sustained , and short-term administration of eltrombopag (25 mg/day) was safe, but it did not effectively improve the patients’ platelet counts.
What is the impact?
If patients clinically diagnosed with immune thrombocytopenia do not respond to immunosuppressive agents, genetic testing should be conducted to exclude hereditary thrombocytopenia, and a normal mean platelet volume should not exclude the possibility of the disease.
For patients with ANKRD26-RT, eltrombopag is safe for short-term use;however, 25 mg/day treatment is unresponsive.
Ourreport complements data on the diagnosis and management of ANKRD26-RT disease in children.
Introduction
ANKRD26-RT is a non-syndromic autosomal dominant thrombocytopenia caused by monoallelic single nucleotide substitutions in the 5’UTR (5’ Untranslated Region) of the ANKRD26 gene mapping to chromosome 10p11.1-p12.Citation1–3 The clinical features include varying increased bleeding propensity, mild to moderate thrombocytopenia with normal mean platelet volume, increased serum TPO (Thrombopoietin) level, impaired bone marrow megakaryopoiesis, and susceptibility to myeloid tumors and other hematological malignancies. In terms of treatment, immunosuppressive treatments are not responsive; however, short-term use of eltrombopag has been reported to safely and effectively improve platelet count.Citation4,Citation5 However, these reports are limited. Therefore, we report the clinical characteristics and treatment efficacy of eltrombopag in two Chinese pediatric patients with the aim to provide data that will assist in the diagnosis and treatment of this heterogeneous disorder.
Patients and methods
Patients
In this study, we enrolled two patients diagnosed with ANKRD26-related thrombocytopenia and eligible for pedigree verification at the Pediatric Department of the First Affiliated Hospital of Guangxi Medical University between January 2019 and December 2020. Clinical characteristics, laboratory tests, gene tests, and therapeutic results were retrospectively analyzed. The exclusion criteria were secondary thrombocytopenia, including thrombocytopenia induced by autoimmune disease, thyroid disease, proliferative diseases of the lymphatic system, myelodysplastic (aplastic anemia and myelodysplastic syndrome), hematologic malignancies, and chronic liver disease hypersplenism, drug-induced thrombocytopenia, alloimmune thrombocytopenia, platelet depletion and secondary thrombocytopenia caused by infections, pseudo-thrombocytopenia, and microangiopathy hemolytic anemia, including acquired and hereditary thrombotic thrombocytopenic purpura. This study was approved by the Ethics Committee of Guangxi Medical University. Informed consent was obtained from both participants’ legal guardians in accordance with the institutional ethics guidelines.
Methods
Data collection
We collected and summarized the clinical data of the two pediatric patients, including the age of onset, sex, clinical presentation, and family history. Laboratory test results before treatment were also collected and analyzed, including the lowest platelet count, white blood cell count, hemoglobin level, mean platelet volume, platelet morphology, and bone marrow cytology. Bleeding tendency was measured using the World Health Organization (WHO) bleeding scale: grade 0, no bleeding; grade 1, petechiae; grade 2, mild blood loss (clinically significant); grade 3, gross blood loss; and grade 4, debilitating blood loss.Citation6
Genetic screening
Peripheral venous blood (5 mL) was collected from the two patients and their respective family members using an ethylenediaminetetraacetic acid (EDTA) anticoagulant tube that was then sent to Beijing Kangxu Medical Laboratory for genetic testing. Mutations in ANKRD26 were detected by high-throughput second-generation sequencing and confirmed by Sanger sequencing. To screen for previously reported mutations, the sequence was compared to reference sequences using HGMD pro, PubMed, Clinvar, and other databases. Single nucleotide polymorphisms (SNPs) were excluded from the ESP, 1000 genome, database and EXAC databases.
Treatment evaluation
We retrospectively analyzed the treatment of two patients with immunosuppressive agents and the thrombopoietin receptor agonist eltrombopag. Both patients were initially diagnosed with immune thrombocytopenia(ITP) and administered immunosuppressive therapy. The type and dose of immunosuppressants, the total course of treatment, platelet counts, and bleeding features at week 4 and throughout the course of treatment were recorded. After immunosuppressive therapy was unresponsive or responded temporarily but could not be maintained and genetic testing indicated mutations in the ANKRD26 gene, the two patients were treated with oral eltrombopag. The dosage and total course of treatment, platelet counts and bleeding features at weeks 4 and 8, and at the end of the course of treatment, were recorded. The observation endpoint of the total treatment course was the end of drug withdrawal or until December 2020. Definitions of response to treatment for the study were as follows: Complete response (CR): A platelet count ≥ 100 × 109/L and the absence of bleeding; Response (R): platelet count ≥ 30 × 109/L and a greater than 2-fold increase in platelet count from baseline and the absence of bleeding; no response (NR): platelet count < 30 × 109/L or less than 2-fold increase in platelet count from baseline or the presence of bleeding.Citation7 Adverse events were graded using CTC-NCI version 5.0.Citation8
Results
Clinical characteristics
Patient 1, a 1-year-old boy, presented with easy bruising (grade 1 WHO bleeding scale). No history of hematological malignancy was reported in the family; however, the father had a history of thrombocytopenia (76–91.5 × 109/L) with no bleeding symptoms. Patient 2, a 1-year-old girl, presented with recurrent hemorrhagic spots(WHO bleeding scale 1). Similar to patient 1, no history of hematological malignancy was reported in the family, but the father had a history of thrombocytopenia (34–82 × 109/L) with no bleeding symptoms ().
Table I. Patients’ clinical characteristics.
Laboratory test results revealed that both patients had a decreased platelet count of less than 50 × 109/L, with normal mean platelet volume, platelet morphology, white blood cell count (the slightly elevated white blood cells in patient1 may be associated with the initial viral infection), and hemoglobin level. Both bone marrow examinations revealed active megakaryocyte proliferation with no obvious impaired maturation. Classified as 50, patient 1 included 2 promegakaryocytes(4%), 36 granular megakaryocytes(72%), 8 thrombocytogenic megakaryocytes(16%) (low platelet production), 4 naked megakaryocytes(8%), and few platelets; patient 2 included 8 promegakaryocytes(16%), 40 granular megakaryocytes(80%), 2 thrombocytogenic megakaryocytes(4%), and a few platelets. ().
Table II. Patients’ laboratory characteristics.
Genetic results
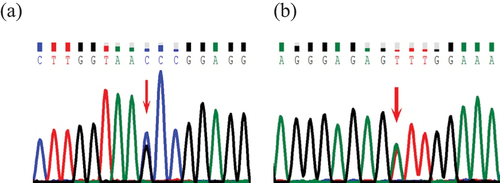
Both patients had mutations in ANKRD26. Patient 1 harbored a previously reported mutation c.-140C>G in the 5’UTR,Citation9,Citation10 inherited from his father. Patient 2 had a mutation c.-127A>T in the 5’UTR. The mutation c.-127A>T, first reported in 2011,Citation11 was inherited from her father. Both fathers had thrombocytopenia with no bleeding symptoms (, ).
Figure 1. Representative chromatograms from direct sequencing of the ANKRD26 gene. The red arrow indicates the mutation position. (a) represents the mutation c.-140C>G of ANKRD26 in 5’UTR in patient 1. (b) represents the mutation c.-127A>T of ANKRD26 in 5’UTR in patients 2.
Treatment results
Both patients were initially diagnosed with ITP and treated with immunosuppressants. Patient1 had a severely low platelet count of 2 × 109/L, which was treated with intravenous immunoglobulin(IVIG), prednisone, dexamethasone, and rituximab for a total observation period of 3 months. Dosage: (1) IVIG: a single dose of 1.0 g/kg/1–2d.; (2) Dexamethasone: 0.6 mg/kg/d for 4 days, intravenously (IV), max40mg/d; Prednisone: initial dose of 1.5 mg/kg/d, dosage adjusted according to platelet count and bleeding symptoms; (3) Rituximab: 100 mg/week for 4 weeks. After treatment with IVIG, dexamethasone, prednisone, and rituximab, the platelet count was 22.4 × 109/L at week 4, and the curative effect was considered unresponsive. After the second dose of rituximab, the platelet count rose briefly to 55 × 109/L, but was not maintained, and decreased to 4.9 × 109/L at week 6. The platelet count fluctuated from 4 × 109/L to 51 × 109/L throughout the 3 months observation period. Thus, the treatment efficacy was temporary response, but was not maintained. Patient 2 had a platelet count of 40 × 109/L before treatment, with recurrent hemorrhagic spots and ecchymosis. Following her family’s wish to initiate treatment, she received 15 months of discontinuous treatment with prednisone, including 7 months of irregular treatment in other hospitals. Dosage: initial dose 1.5 mg/kg/d, dosage adjusted according to platelet count and bleeding symptoms. After 4 weeks of treatment, the platelet count was 60 × 109/L, which fluctuated from 40 × 109/L to 60 × 109/L throughout the 7 months of treatment. After 2 months of complete prednisone cessation, the platelet count dropped to 34 × 109/L, recurrent hemorrhagic spots and ecchymosis appeared, and her family came to our hospital. Oral prednisone therapy was administered again for 6 months, during which the platelet count fluctuated from 34 × 109/L to 72 × 109/L. No curative effect was observed (, ). Adverse events: All patients received prophylactic gastrointestinal treatment and calcium supplementation to prevent osteoporosis during corticosteroid treatment. Patient 1 developed bronchopneumonia and fungal infection (CTC-NCI grade 3) during rituximab treatment, and no serum sickness was observed. A cushingoid appearance (CTC-NCI grade 1) was observed during treatment with dexamethasone and prednisone. Patient 2 developed a cushingoid appearance (CTC-NCI grade 1) during the prednisone treatment.
Figure 2. (a) Therapeutic effect of immunosuppressor in patient 1. (b) Therapeutic effect of immunosuppressor in patient 2.
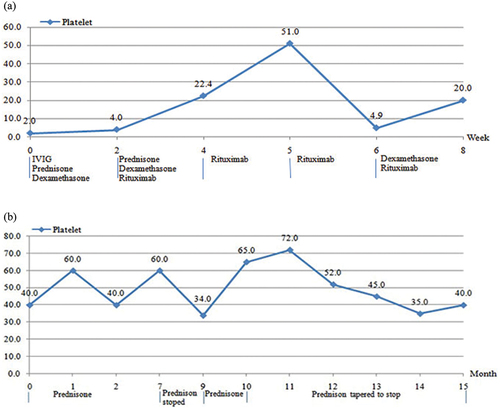
Table III. Therapeutic effect of immunosuppressor.
After the failure of immunosuppressive therapy and genetic testing revealed ANKRD26 gene mutations, both patients were administered oral thrombopoietin receptor agonist eltrombopag(25 mg/d). The total course of treatment for patient 1 was 2 months, with a platelet count of 29.8 × 109 /L before treatment, 18 × 109 /L and 11.3 × 109 /L at 4 and 8 weeks of treatment, respectively, showing no increase compared to the pre-treatment period. The total curative effect was considered no responsive, with no adverse events. After eltrombopag was discontinued, the platelet count was no longer monitored, and no bleeding symptoms were observed during follow-up. The patient 2 family decided to stop the administration of eltrombopag after 1 month of treatment, and the platelet count observed subsequently only increased from 40 × 109/L to 55 × 109/L after treatment, showing an unresponsive curative effect. No adverse events were observed during the treatment period, except for a small rash that resolved on its own(CTC-NCI grade 1). After discontinuation of the drug, the platelet count was monitored irregularly and fluctuated from 32 × 109 to 50 × 109/L. No bleeding symptoms were observed ().
Table IV. Therapeutic effect of eltrombopag.
Discussion
ANKRD26-RT is a rare autosomal dominant disorder with no significant geographic or sex differences in its onset.Citation11 We studied two Chinese pediatric cases, a boy and a girl, aged 1, and diagnosed with ANKRD26-RT and low platelet count of <50 × 109/L, the boy, had a platelet count as low as <10 × 109/L. Both presented with slight bleeding classified as WHO score 1, and they had not experienced any serious life-threatening bleeding events, which was consistent with previous reports.Citation9–12 A clinical feature of this disease that has previously attracted attention is the increased risk of hematological malignancies, especially myeloid tumors. In this study, both patients and their family members had no history of hematologic malignancies; however, further follow-up is required because both patients were still very young. Apart from a normal white blood cell count, hemoglobin level, and platelet morphology, both patients had a normal mean platelet volume, which can potentially be a nonspecific distinguisher from some hereditary thrombocytopenia with abnormal platelet volume. Previous reports have shown that the platelet count is temporarily normalized during viral infection.Citation10,Citation11 However, this was not observed in patient 1, who had a viral infection at the beginning of the disease. However, his platelet count severely decreased, and whether this was related to the specific infectious pathogen remains unclear. It has been reported that bone marrow usually showed increased megakaryocytes and dysmegakaryopoiesis, and dystrophic forms consisted mainly of small, underdeveloped megakaryocytes and typical micromegakaryocyte.Citation1,Citation10,Citation11,Citation13 Bone marrow examination of the two patients mainly revealed active megakaryocyte proliferation with no obvious impaired maturation. As there is no specific clinical manifestation and laboratory examinations for the differential diagnosis of ITP, it has been proposed that ANKRD26-RT should be suspected in individuals with the following: (1) lifelong mild to moderate thrombocytopenia (<150 × 109/L, confirmed with repeated examinations); (2) normal platelet size (mean platelet volume [fL] per reference interval of automated instrument); (3) absent or minimal bleeding tendency; (4) family history of thrombocytopenia with an autosomal dominant pattern of inheritance; (5) personal or family history of myeloid neoplasms at a young age; (6) previous or suspected diagnosis of immune thrombocytopenia (ITP) without improvement of immunosuppressive treatment; and (7) absence of features suggesting syndromic association.Citation14 Finally, mutant molecular confirmation was performed to establish the diagnosis. Nevertheless, the mutations are sometimes difficult to identify, especially if the disease is mainly characterized by mild to moderate platelet reduction with no or slight bleeding, resulting in clinicians missing the inheritance source and correlation with the family history of thrombocytopenia. The main features that prompted our molecular examination were isolated thrombocytopenia, a normal platelet volume, and no response to immunosuppressive therapy or temporary response, but not sustained. We believe that these indicators suggest the need for genetic screening.
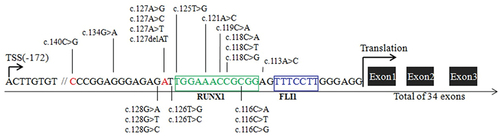
ANKRD26 is the ancestor of a family of primate-specific genes termed POTE,Citation15 With regard to human bone marrow cells, ANKRD26 is expressed in megakaryocytes, and, to a lesser extent, in erythroid cells.Citation1,Citation16 Thrombocytopenia 2 (THC2) disorder, first described in two large families in Italy and North America, has a pathogenic gene locus located on the short arm of chromosome 10 (10p11.1-P12).Citation2,Citation3 After Pippucci et al.Citation1 revealed that THC2 was not caused by MASTL or ACBD5 defects, but was rather associated with ANKRD26 mutations, subsequent studies have reported that the specific pathogenesis is mainly due to the 5’UTR mutation of the ANKRD26 gene. This mutation has been shown to lead to the loss of RUNX1and FLI1 binding at the 5’UTR, resulting in the continued overexpression of ANKRD26, leading to abnormal activation of the ERK/MAPK pathway and impaired megakaryopoiesis.Citation17,Citation18 Currently, 11 single nucleotide mutations(c.-113,c.-116,c.-118,c.-119, c.-121,c.-125,c.-126,c.-127,c.-128,c.-134, and c.-140) involving 21 mutation types within the 5′ UTR mutation of ANKRD26 have been confirmed to be pathogenic, with the most common type being c.-128 G>A, as reported from a total of 84 individuals from 19 families to date ().Citation1,Citation10,Citation11,Citation19–26 In addition, a missense mutation c. 473A>G in the ANKRD26 exon region has been reported to segregate with thrombocytopeniaCitation27; however, it has been proposed that this may not be the real pathogenic mutation, as this disease is caused by failing to silence ANKRD26 transcription rather than loss of protein function.Citation25 function
Figure 3. Representative mutations in 5’UTR of the ANKRD26 gene associated with ANKRD26-RT. Position c-172 indicates the transcription start site (TSS). Green and blue areas represent RUNX1 and FLI1 binding sites. The red position represents the location of the mutation in our patient.
In this study, the final diagnosis in our two patients was established after mutations in ANKRD26were detected by genetic screening. Two mutations inherited from the parents were identified, namely ANKRD26 c.-140C>G in patient 1 and ANKRD26 c.-127A>T in patient 2. Mutations c.-140C>G and c.-127A>T have been previously reported to lead to thrombocytopenia, and the two fathers carrying the heterozygous mutation in this study had thrombocytopenia, which is consistent with theANKRD26-RT diagnosis.
Immunosuppressant therapy has been reported to be a no response or temporary increase but is ultimately unable to maintain platelet stability.Citation10,Citation11,Citation13 In this study, our two patients were initially diagnosed with ITP and treated with immunosuppressants for 3 and 15 months, respectively. Patient 2 had a platelet count of 40 × 109/L before treatment, with recurrent hemorrhagic spots and ecchymosis. The decision to treat relies largely on the frequency and severity of bleeding and the impact on quality of lifeCitation7. Bleeding risk appears to increase when platelets are <20 × 109/L, while patient 2 had a platelet count of >30 × 109/L before treatment, with mild bleeding and no specific needs such as surgery. Considering the degree of bleeding, we can choose to observe rather than treat it. However, she had recurrent hemorrhagic spots and ecchymosis, which affected the child’s quality of life, and the family was anxious about this and strongly desired treatment. Considering the preference of the patient’s family, we chose to treat patient 2. A review of the entire course of treatment showed that immunosuppressive treatment was no response at week 4 and throughout the course of treatment in patient 2. For patient 1, the treatment was temporary response, but was not sustained. As immunosuppressants have certain toxic side effects, an accurate diagnosis would help choose the correct treatment plan which and avoid the excessive use of immunosuppressants.
Patients with hereditary thrombocytopenia usually require transfusion of platelets to improve platelet counts before surgery or other invasive procedures as well as in cases of significant spontaneous bleeding. The use of platelet receptor agonists helps to avoid the risks associated with platelet transfusion, such as exposure to acute reactions, transmission of infectious diseases, alloimmunity, and blood shortage. A previous study has reported that the platelet count in an adult patient with ANKRD26-RT can be successfully improved by short-term preoperative administration of eltrombopag at a dose of 50–75 mg/d instead of platelet transfusion.Citation4 A phase II clinical trial has revealed that eltrombopag can safely and effectively increase the platelet count in patient with ANKRD26-RT, even though it was still observed that the degree of platelet response in patient with ANKRD26-RT was generally lower than that in other types of hereditary thrombocytopenia, such as MYH9 related and mBSS related thrombocytopenia. Most participants in the trial had a mild response (platelets were at least 2 times higher than baseline), and for most of those who did not have this response, the platelet count later increased after increasing the dose to 75 mg/d for 3 weeks.Citation5 In our study, the two patients were administered oral eltrombopag at a dose of 25 mg/day for 8 and 4 weeks, respectively, but the treatment was no response. To evaluate the association of the degree of treatment response with eltrombopag dose, further clinical studies are needed. Patients with ANKRD26-RT are at risk of myeloid tumors, and the TPO/MPL pathway can be activated by thrombopoietin receptor agonists. Thus, the association risk of probable disease progression to myeloid tumors remains a major concern. No adverse events were observed in our two patients during the course of treatment, except for a small rash(CTC-NCI grade 1) that faded on its own in patient 2, with no other blood changes except for platelet count. This is consistent with the previous reports that the short-term use of eltrombopag is safe. However, the overall course of treatment in this study was not long and the number of cases was small. The safety of long-term eltrombopag use requires further investigation.
In conclusion, we report two Chinese pediatric patients with ANKRD26-RT who initially presented with mild bleeding accompanied by different degrees of platelet reduction, normal mean platelet volume, and megakaryocyte maturation impairment not obvious. The normal mean platelet volume can be distinguished by ANKRD26-RT from other hereditary thrombocytopenia with abnormal platelet volume; however, no other laboratory tests revealed differential markers. If patients clinically diagnosed with ITP do not respond to immunosuppressive agents, genetic testing is necessary to exclude hereditary thrombocytopenia. The short-term use of eltrombopag at a dose of 25 mg/day failed to effectively improve the platelet count for our two patients. However, the association of the degree of treatment response with dose was not characterized in the study. For patients with ANKRD26-RT, eltrombopag is safe for short-term use; however, its long-term effects and safety remain unclear. Our report complements data on the diagnosis and management of ANKRD26-RT disease in children.
Acknowledgments
We are grateful to the two patients and their families for their participation in the study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Pippucci T, Savoia A, Perrotta S, Pujol-Moix N, Noris P, Castegnaro G, Pecci A, Gnan C, Punzo F, Marconi C, et al. Mutations in the 5′ UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am JHum Genet. 2011;88:115–7. doi:10.1016/j.ajhg.2010.12.006.
- Savoia A, Del Vecchio M, Totaro A, Perrotta S, Amendola G, Moretti A, Zelante L, Iolascon A. An autosomal dominant thrombocytopenia gene maps to chromosomal region 10p. Am J Hum Genet. 1999;65(5):1401–5. doi:10.1086/302637.
- Drachman JG, Jarvik GP, Mehaffey MG. Autosomal dominant thrombocytopenia: incomplete megakaryocyte differentiation and linkage to human chromosome 10. Blood. 2000;96(1):118–25. doi:10.1182/blood.V96.1.118.
- Fiore M, Saut N, Alessi MC, Viallard JF. Successful use of eltrombopag for surgical preparation in a patient with ANKRD26-related thrombocytopenia. Platelets. 2016;27(8):828–9. doi:10.1080/09537104.2016.1190446.
- Zaninetti C, Gresele P, Bertomoro A, Klersy C, De Candia E, Veneri D, Barozzi S, Fierro T, Alberelli MA, Musella V, et al. Eltrombopag for the treatment of inherited thrombocytopenias: a phase II clinical trial. Haematologica. 2020;105(3):820–8. doi:10.3324/haematol.2019.223966.
- Fogarty PF, Tarantino MD, Signorovitch J, Grotzinger KM. Selective validation of the WHO bleeding scale in patients with chronic immune thrombocytopenia. Curr Med Res Opin. 2012;28:79–87. doi:10.1185/03007995.2011.644849.
- Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Crowther MA, American Society of Hematology. The American society of hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117:4190–207. doi:10.1182/blood-2010-08-302984.
- NCI. National cancer institute common terminology criteria for adverse events (CTCAE) v5.0. [Accessed 2018 Nov 8]. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm.2017.
- Ferrari S, Lombardi AM, Putti MC. Spectrum of 5‘UTR mutations in ANKRD26 gene in patients with inherited thrombocytopenia: c.-140C>G mutation is more frequent than expected. Platelets. 2017;28:621–4. doi:10.1080/09537104.2016.1267337.
- Boutroux H, Petit A, Auvrignon A, Lapillonne H, Ballerini P, Favier R, Leverger G. Childhood diagnosis of genetic thrombocytopenia with mutation in the ankyrine repeat domain 26 gene. EurJPediatr. 2015;174(10):1399–403. doi:10.1007/s00431-015-2549-x.
- Noris P, Perrotta S, Seri M, Pecci A, Gnan C, Loffredo G, Pujol-Moix N, Zecca M, Scognamiglio F, De Rocco D, et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: analysis of 78 patients from 21 families. Blood. 2011;117(24):6673–80. doi:10.1182/blood-2011-02-336537.
- Noris P, Pecci A. Hereditary thrombocytopenias: a growing list of disorders. Hematology Am Soc Hematol Educ Program. 2017;2017(1):385–99. doi:10.1182/asheducation-2017.1.385.
- Zaninetti C, Santini V, Tiniakou M, Barozzi S, Savoia A, Pecci A. Inherited thrombocytopenia caused by ANKRD26 mutations misdiagnosed and treated as myelodysplastic syndrome: report on two cases. J Thromb Haemost. 2017;15(12):2388–92. doi:10.1111/jth.13855.
- Perez BJ, Dugan SN, Anderson MW. ANKRD26-related thrombocytopenia. In: Adam M, Mirzaa G, Pagon R, Wallace S, Bean L, Gripp K Amemiya A, editors. GeneReviews®[Internet]. Seattle (WA): University of Washington; 2018. p. 1993–2022.
- Hahn Y, Bera TK, Pastan IH, Lee B. Duplication and extensive remodeling shaped POTE family genes encoding proteins containing ankyrin repeat and coiled coil domains. Gene. 2006;366(2):238–45. doi:10.1016/j.gene.2005.07.045.
- Macaulay IC, Tijssen MR, Thijssen-Timmer DC, Gusnanto A, Steward M, Burns P, Langford CF, Ellis PD, Dudbridge F, Zwaginga JJ, et al. Comparative gene expression profiling of in vitro differentiated megakaryocytes and erythroblasts identifies novel activatory and inhibitory platelet membrane proteins. Blood. 2007;109(8):3260–9. doi:10.1182/blood-2006-07-036269.
- Bluteau D, Balduini A, Balayn N, Currao M, Nurden P, Deswarte C, Leverger G, Noris P, Perrotta S, Solary E, et al. Thrombocytopenia associated mutations in the ANKRD26 regulatory region induce MAPK hyperactivation. J Clin Invest. 2014;124(2):580–91. doi:10.1172/JCI71861.
- Balduini A, Raslova H, Di Buduo CA, Donada A, Ballmaier M, Germeshausen M, Balduini CL. Clinic, pathogenic mechanisms and drug testing of two inherited thrombocytopenias, ANKRD26-related thrombocytopenia and MYH9-related diseases. Eur J Med Genet. 2018;61(11):715–22. doi:10.1016/j.ejmg.2018.01.014.
- Noris P, Favier R, Alessi MC, Geddis AE, Kunishima S, Heller PG, Giordano P, Niederhoffer KY, Bussel JB, Podda GM, et al. ANKRD26-related thrombocytopenia and myeloid malignancies. Blood. 2013;122(11):1987–9. doi:10.1182/blood-2013-04-499319.
- Diep RT, Corey K, Arcasoy MO. A novel nucleotide substitution in the 5′untranslated region of ANKRD26 gene is associated with inherited thrombocytopenia: a report of two new families. Ann Hematol. 2019;98(7):1789–91. doi:10.1007/s00277-019-03632-y.
- Ventz R, Hundemer M, Witzens-Harig M, Lehmann B, Felbor U, Najm J. Mild bleeding diathesis in a 62-year-old woman with hereditary thrombocytopenia. Internist (Berl). 2013;54(6):765–8. doi:10.1007/s00108-013-3284-x.
- Guison J, Blaison G, Stoica O, Hurstel R, Favier M, Favier R. Idiopathic pulmonary embolism in a case of severe family ANKRD26thrombocytopenia. Mediterr J Hematol Infect Dis. 2017;9(1):e2017038. doi:10.4084/mjhid.2017.038.
- Averina M, Jensvoll H, Strand H, Sovershaev M. A novel ANKRD26 gene variant causing inherited thrombocytopenia in a family of Finnish origin: another brick in the wall? Thromb Res. 2017;151:41–3. doi:10.1016/j.thromres.2017.01.001.
- Fournier E, Debord C, Soenen V, Sovershaev M. Baseline dysmegakaryopoiesis in inherited thrombocytopenia/platelet disorder with predisposition to haematological malignancies. Br J Haematol. 2020;189(4):e119–e22. doi:10.1111/bjh.16543.
- Tan C, Dai L, Chen Z, Yang W, Wang Y, Zeng C, Xiang Z, Wang X, Zhang X, Ran Q, et al. A rare big Chinese family with thrombocytopenia 2: a case report and literature review. Front Genet. 2020;11:340. doi:10.3389/fgene.2020.00340.
- Zidan NI, AbdElmonem DM, Elsheikh HM, Metwally EA, Mokhtar WA, Osman GM. Relation between mutations in the 5’ UTR of ANKRD26 gene and inherited thrombocytopenia with predisposition to myeloid malignancies. An Egyptian study. Platelets. 2021;32:642–50. doi:10.1080/09537104.2020.1790512.
- Al Daama SA, Housawi YH, Dridi W, Sager M, Otieno FG, Hou C, Vasquez L, Kim C, Tian L, Sleiman P, et al. A missense mutation in ANKRD26 segregates with thrombocytopenia. Blood. 2013;122(3):461–2. doi:10.1182/blood-2013-03-489344.