
While mouse models have proven to be valuable tools for studying human diseases, mouse models also have certain limitations that need to be considered. Some of the genetic differences between mice and humans are derived from differences in coding sequences, which give rise to proteins with different properties. For example, mouse hemoglobin has a lower affinity than human hemoglobin for, and the lower affinity for O2 facilitates the dissociation of O2 from hemoglobin in peripheral tissues and helps to support the higher metabolic rate in mice.Citation1 Researchers are familiar with the larger number of thrombocytes in mice than in humans, but a reasonable explanation for the difference has not been proposed.
Hereditary thrombocytosis (HT) with autosomal-dominant transmission has been described with manifestations similar to those of sporadic essential thrombocythemia. Germline mutation of the thrombopoietin gene (THPO),Citation2–9 the thrombopoietin receptor gene (MPL)Citation10–15 or the JAK2 geneCitation16 was reported as the cause of human HT. THPO and MPL are the key regulators for stem cell proliferation and the terminal differentiation of platelets.Citation17 For all of the THPO mutations identified in HT, it was reported that the mutations caused increased translational efficiency of THPO mRNA with overproduction of normal thrombopoietin molecules and stimulation of the production of platelets. Mutation of MPL in the trans-membrane region (MPL S505N) causes constitutive activation of the receptor independently of the binding of thrombopoietin.Citation18 Mutations of MPL involving cytokine receptor motifs of the extracellular portion (MPL K39N and P106L) caused low binding affinity of MPL to thrombopoietin, resulting in high thrombopoietin levels.Citation18
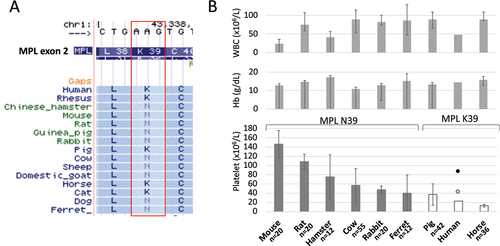
We hypothesized that the relatively higher platelet count in mice is caused by the genetic background shared in the species. First, we examined the reported germline THPO mutations including THPO c.-47delG,Citation2 c.-31 G>T,Citation3 c.13 G>A,Citation4 c.13 + 1 G>C,Citation5 c.13 + 2T>C,Citation6 and c.13 + 5 G>A,Citation8 but we did not find any variants in murine Thpo. We also checked the mouse Thpo transcript associated with exon 3 skipping, which is a mechanism shared in human HT,Citation5–7 but we could not identify an exon 3 skipping transcript (data not shown). We then focused on somatic MPL W515L/KCitation19 and JAK2 V617F mutations reported in myeloproliferative neoplasms and germline MPL variants (K39N,Citation13 R102P,Citation20 P106L,Citation14 R120W,Citation21 R170H,Citation21 T487A,Citation22 L502G,Citation23 S505N)Citation15 and JAK2 variants (I223T, R564Q, H608N, V617I, S755R, R867Q, T875N, R938Q)Citation16,Citation24 reported in human HT. None of the JAK2 variants reported in humans were found in mouse proteins. On the other hand, MPL protein showed differences between mice and humans at three (K39N, R120W, T487A) of the nine MPL variant sites reported in human HT and myeloproliferative neoplasm (). Of those three amino acid loci, mice have Mpl N39, instead of MPL K39 human reference, which is identical to the K39N variant reported in human HT (). We confirmed the variant in commonly used mouse strains including C57BL/6, BALB/c, DBA/2, and C3H strains, and they all showed MPL N39 (data not shown).
Figure 1. Comparison of human and mice MPL proteins.
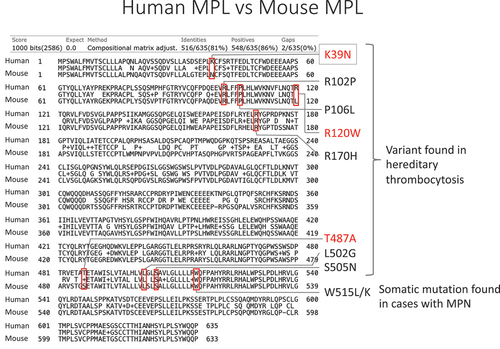
Human MPL (635aa) and mouse Mpl (633aa) were compared in the alignment function of BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Human MPL and mouse MPL showed 81% identities. The red boxes indicate the known MPL variants in human hereditary thrombocytosis or myeloproliferative neoplasm (MPN).
MPL K39N in humans, also known as MPL Baltimore, results from the substitution of lysine with asparagine at amino acid 39. The mutation was first identified in three African Americans, and about 7% of African Americans are heterozygous for this mutation.Citation13 This polymorphism is autosomal and appears to conform to a pattern of autosomal dominance with incomplete penetrance, considering that some heterozygotes have normal platelet counts and others have sustained elevations. The mutation in the homozygous state is associated with severe thrombocytosis.Citation18 Mpl K39N located at the cytokine receptor motifs of the extracellular portion results in low binding affinity of MPL to thrombopoietin. Moreover, due to a glycosylation defect, MPL expression is impaired, and thrombopoietin clearance is reduced in this hereditary thrombocytosis.Citation18 Mouse and some other species share the MPL N39 variant (). These species are thought to have the homozygous Mpl variant if there is no Mpl N/K39 polymorphism in the species. A comparison of complete blood counts in mammalsCitation25 showed that the Mpl K39N variant was shared among mammals, which showed relative thrombocytosis compared to that in humans (). Mean platelet counts in mammalian species with Mpl N39 were higher than those in mammalian species with Mpl K39 (P = .024, Mann–Whitney U test). Moreover, human subjects with homozygous MPL N39 showed higher platelets counts (mean platelet count: 858,000/µL) than those in human subjects with heterozygous MPL N39 (mean platelet count: 424,000/µL) and those in human subjects with MPL K39 in a general population ().Citation13 There are still large variations of platelet counts among species with Mpl N39. An increased number of thrombocytes might have been advantageous for the ancestors of some species to stop bleeding or it might just have been a bystander founder effect of those species. A comparison between species is difficult because it is like a comparison between apples and oranges. However, the results of comparative genomic analysis showed that mammalian species with Mpl N39 had significantly higher platelet counts than those in mammalian species with Mpl K39. Of note, the variant is known as a human polymorphism that contributes to human HT.
Figure 2. a) Vertebrate alignment track of UCSC genome browser (http://genome.Ucsc.edu/cgi-bin/hgGateway) showed the variation of MPL K/N39 among mammalian species. B) Comparison of blood counts in mammals. Species are sorted by the higher average platelet count from left to right. Compared to level of WBC and Hb, which are comparable among species, platelets showed a great diversity among species. Species with MPL N39 showed higher platelet counts than those in species with MPL K39. The error bar represents standard deviation. The filled circle indicates the mean platelet count of individuals with homozygous K39N. The blank circle indicates mean platelet count of individuals with heterozygous K39N. Mammalian blood count data were objected from Shiraishi J et al.Citation25 Mean platelet counts of individuals with MPL K39N were objected from Moliterno AR et al.Citation13
Additional supporting clues of the contribution of Mpl K39 to relative thrombocytosis have been reported.Citation26,Citation27 Mice with an adjacent Mpl C40R mutation showed relative thrombocytopenia.Citation26 Transgenic mice with variant MPL splicing, by which seven adjacent amino acids (V28-D34) are eliminated, also showed lower platelet counts than those in wild-type mice.Citation27 These findings support the notion that the region plays an important role in the maintenance of thrombocytosis in mouse.
Mice and rats have frequently been used as animal models recapitulating human diseases; however, we need to be careful in interpreting the results. Enforced expression of THPO in mice results in the development of thrombocytosis and myelofibrosis.Citation28,Citation29 On the other hand, individuals with hereditary thrombocytosis caused by THPO mutation rarely develop myelofibrosis. Similarly, a CRISPR/Cas9-engineered mouse model harboring a germline Mpl S504N mutation developed essential thrombocythemia and myelofibrosis,Citation30 whereas the initial pedigree with MPL S505N mutation showed thrombocytosis without myelofibrosis.Citation15 These differences might be due to the different genetic backgrounds of species. Mice could be genetically preconditioned to have an enhanced signal of the Thpo-Mpl axis due to homozygous Mpl N39, thus making them prone to develop myelofibrosis compared to humans. It is essential to consider these species-specific differences when studying platelet biology or using mouse models to investigate platelet-related disorders.
Supplemental Material
Download PDF (213.2 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/09537104.2023.2276697
References
- Perlman RL. Mouse models of human disease: an evolutionary perspective. Evol Med Public Health. 2016;2016(1):170–4. doi:10.1093/emph/eow014.
- Kondo T, Okabe M, Sanada M, Kurosawa M, Suzuki S, Kobayashi M, Hosokawa M, Asaka M. Familial essential thrombocythemia associated with one-base deletion in the 5′-untranslated region of the thrombopoietin gene. Blood. 1998;92(4):1091–6. doi:10.1182/blood.V92.4.1091.
- Kikuchi M, Tayama T, Hayakawa H, Takahashi I, Hoshino H, Ohsaka A. Familial thrombocytosis. Br J Haematol. 1995;89(4):900–2. doi:10.1111/j.1365-2141.1995.tb08432.x.
- Prouzet-Mauleon V, Montibus B, Chauveau A, Hautin M, Migeon M, Ka C, Laharanne E, Bidet A, Corcos L, Lippert E, et al. A novel thrombopoietin (THPO) mutation altering mRNA splicing in a case of familial thrombocytosis. Br J Haematol. 2020;190(2):e104–e7. doi:10.1111/bjh.16742.
- Wiestner A, Schlemper RJ, van der Maas AP, Skoda RC. An activating splice donor mutation in the thrombopoietin gene causes hereditary thrombocythaemia. Nat Genet. 1998;18(1):49–52. doi:10.1038/ng0198-49.
- Zhang B, Ng D, Jones C, Oh ST, Nolan GP, Salehi S, Wong W, Zehnder JL, Gotlib J. A novel splice donor mutation in the thrombopoietin gene leads to exon 2 skipping in a Filipino family with hereditary thrombocythemia. Blood. 2011;118(26):6988–90. doi:10.1182/blood-2011-10-386177.
- Liu K, Kralovics R, Rudzki Z, Grabowska B, Buser AS, Olcaydu D, Gisslinger H, Tiedt R, Frank P, Okon K, et al. A de novo splice donor mutation in the thrombopoietin gene causes hereditary thrombocythemia in a Polish family. Haematologica. 2008;93(5):706–14. doi:10.3324/haematol.11801.
- Jorgensen MJ, Raskind WH, Wolff JF, Bachrach HR, Kaushansky K. Familial thrombocytosis associated with overproduction of thrombopoietin due to a novel splice donor site mutation. Blood (ASH Meeting Abstracts). 1998;92:Abstract #834.
- Harrison CN, Gale RE, Wiestner AC, Skoda RC, Linch DC. The activating splice mutation in intron 3 of the thrombopoietin gene is not found in patients with non-familial essential thrombocythaemia. Br J Haematol. 1998;102(5):1341–3. doi:10.1111/j.1365-2141.1998.00921.x.
- Ding J, Komatsu H, Iida S, Yano H, Kusumoto S, Inagaki A, Mori F, Ri M, Ito A, Wakita A, et al. The Asn505 mutation of the c-MPL gene, which causes familial essential thrombocythemia, induces autonomous homodimerization of the c-Mpl protein due to strong amino acid polarity. Blood. 2009;114(15):3325–8. doi:10.1182/blood-2008-04-149047.
- Liu K, Martini M, Rocca B, Amos CI, Teofili L, Giona F, Ding J, Komatsu H, Larocca LM, Skoda RC, et al. Evidence for a founder effect of the MPL-S505N mutation in eight Italian pedigrees with hereditary thrombocythemia. Haematologica. 2009;94(10):1368–74. doi:10.3324/haematol.2009.005918.
- Teofili L, Giona F, Torti L, Cenci T, Ricerca BM, Rumi C, Nunes V, Foa R, Leone G, Martini M, et al. Hereditary thrombocytosis caused by MPLSer505Asn is associated with a high thrombotic risk, splenomegaly and progression to bone marrow fibrosis. Haematologica. 2010;95(1):65–70. doi:10.3324/haematol.2009.007542.
- Moliterno AR, Williams DM, Gutierrez-Alamillo LI, Salvatori R, Ingersoll RG, Spivak JL. Mpl Baltimore: a thrombopoietin receptor polymorphism associated with thrombocytosis. Proc Natl Acad Sci USA. 2004;101(31):11444–7. doi:10.1073/pnas.0404241101.
- El-Harith el HA, Roesl C, Ballmaier M, Germeshausen M, Frye‐Boukhriss H, Von Neuhoff N, Becker C, Nürnberg G, Nürnberg P, Ahmed MAM, et al. Familial thrombocytosis caused by the novel germ-line mutation p.Pro106Leu in the MPL gene. Br J Haematol. 2009;144(2):185–94. doi:10.1111/j.1365-2141.2008.07430.x.
- Ding J, Komatsu H, Wakita A, Kato-Uranishi M, Ito M, Satoh A, Tsuboi K, Nitta M, Miyazaki H, Iida S, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood. 2004;103(11):4198–200. doi:10.1182/blood-2003-10-3471.
- Han EY, Catherwood M, McMullin MF. Hereditary thrombocytosis: the genetic landscape. Br J Haematol. 2021;194(6):1098–105. doi:10.1111/bjh.17741.
- Kaushansky K, Wood AJJ. Thrombopoietin. N Engl J Med. 1998;339(11):746–54. doi:10.1056/NEJM199809103391107.
- Teofili L, Larocca LM. Advances in understanding the pathogenesis of familial thrombocythaemia. Br J Haematol. 2011;152(6):701–12. doi:10.1111/j.1365-2141.2010.08500.x.
- Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, Cuker A, Wernig G, Moore S, Galinsky I, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. doi:10.1371/journal.pmed.0030270.
- Bellanne-Chantelot C, Mosca M, Marty C, Favier R, Vainchenker W, Plo I. Identification of MPL R102P mutation in hereditary thrombocytosis. Front Endocrinol (Lausanne). 2017;8:235. doi:10.3389/fendo.2017.00235.
- Al-Harbi T, Al-Zahrani M, Al-Balwi M, Al‐Hazmi A, Alsuhaibani A, Aljafn N, Alsumari F, Aleshaiwi L, Alsuhibani A, Alqasim O, et al. Clinical course of myeloproliferative leukaemia virus oncogene (MPL) mutation-associated familial thrombocytosis: a review of 64 paediatric and adult patients. Br J Haematol. 2021;194(5):893–8. doi:10.1111/bjh.17624.
- Vasseur L, Favier R, Kim R, Rabian F, Cabannes‐Hamy A, Cassinat B, Maslah N, Vasquez N, Clappier E, Kiladjian J-J, et al. Clonal evolution in hereditary thrombocytosis with MPL T487A mutation. Pediatr Blood Cancer. 2023;70(2):e29905. doi:10.1002/pbc.29905.
- Rendo M, Cavacece C, Kou CJ, Beeler BW, Fenderson J. Familial essential thrombocythemia with novel MPL L502G and G208K mutations. Cureus. 2022;14(3):e23220. doi:10.7759/cureus.23220.
- Muller J, Porret NA, Rufer A. Identification of a JAK2 FERM domain variant associated with hereditary thrombocytosis. Hemasphere. 2021;5(8):e626. doi:10.1097/HS9.0000000000000626.
- Shiraishi J, Matsumoto H, Hirayama H. Experimental blood cell counting on several kinds of animals with an automated hematology analyzer. Sysmex J Int. 2007;17:1–7.
- Chan ER, Lavender H, Li G, Haviernik P, Bunting KD, Adams MD. An ENU-induced recessive mutation in Mpl leads to thrombocytopenia with overdominance. Exp Hematol. 2009;37(2):276–84. doi:10.1016/j.exphem.2008.10.005.
- Spivak JL, Merchant A, Williams DM, Rogers O, Zhao W, Duffield A, Resar LS, Moliterno AR, Zhao ZJ. Thrombopoietin is required for full phenotype expression in a JAK2V617F transgenic mouse model of polycythemia vera. PLoS One. 2020;15(6):e0232801. doi:10.1371/journal.pone.0232801.
- Kakumitsu H, Kamezaki K, Shimoda K, Karube K, Haro T, Numata A, Shide K, Matsuda T, Oshima K, Harada M, et al. Transgenic mice overexpressing murine thrombopoietin develop myelofibrosis and osteosclerosis. Leuk Res. 2005;29(7):761–9. doi:10.1016/j.leukres.2004.12.009.
- Frey BM, Rafii S, Teterson M, Eaton D, Crystal RG, Moore MA. Adenovector-mediated expression of human thrombopoietin cDNA in immune-compromised mice: insights into the pathophysiology of osteomyelofibrosis. J Immunol. 1998;160(2):691–9. doi:10.4049/jimmunol.160.2.691.
- Adriaanse FRS, Kamens JL, Vogel P, Sakurada SM, Pruett-Miller SM, Stam RW, Michel Zwaan C, Gruber TA. A CRISPR/Cas9 engineered Mpl(S504N) mouse model recapitulates human myelofibrosis. Leukemia. 2022;36(10):2535–8. doi:10.1038/s41375-022-01684-0.