
Abstract
Kappa-carrageenan (KCG), which is used to induce thrombosis in laboratory animals for antithrombotic drug screening, can trigger platelet aggregation. However, the cell-surface receptor and related signaling pathways remain unclear. In this study, we investigated the molecular basis of KCG-induced platelet activation using light-transmittance aggregometry, flow cytometry, western blotting, and surface plasmon resonance assays using platelets from platelet receptor-deficient mice and recombinant proteins. KCG-induced tail thrombosis was also evaluated in mice lacking the platelet receptor. We found that KCG induces platelet aggregation with α-granule secretion, activated integrin αIIbβ3, and phosphatidylserine exposure. As this aggregation was significantly inhibited by the Src family kinase inhibitor and spleen tyrosine kinase (Syk) inhibitor, a tyrosine kinase-dependent pathway is required. Platelets exposed to KCG exhibited intracellular tyrosine phosphorylation of Syk, linker activated T cells, and phospholipase C gamma 2. KCG-induced platelet aggregation was abolished in platelets from C-type lectin-like receptor-2 (CLEC-2)-deficient mice, but not in platelets pre-treated with glycoprotein VI-blocking antibody, JAQ1. Surface plasmon resonance assays showed a direct association between murine/human recombinant CLEC-2 and KCG. KCG-induced thrombosis and thrombocytopenia were significantly inhibited in CLEC-2-deficient mice. Our findings show that KCG induces platelet activation via CLEC-2.
Plain Language Summary
Thrombosis is a serious medical condition that occurs when blood clots form in the blood vessels and can lead to heart attacks or strokes. Animal models are important for evaluating the effectiveness of drugs in thrombosis treatment. Kappa-carrageenan (KCG) is a food thickener and a substance used to induce clot formation in laboratory animals. In this study, we investigated the molecular basis of KCG-induced platelet activation, which is an important step in thrombosis development. We found that KCG activates platelets via a receptor called C-type lectin-like receptor 2 (CLEC-2), leading to a prothrombotic state in mice. We also showed that KCG-induced tail thrombosis (CTT) is significantly inhibited in CLEC-2 deficient mice. Our findings suggest that CLEC-2-mediated platelet activation plays a key role in the pathogenesis of thrombosis and CLEC-2 May participate in innate immunity as a receptor for sulfate-polysaccharide.
Abbreviation; CLEC-2: C-type lectin-like receptor 2; CRP: collagen-related peptide; CTT: KCGN-induced tail thrombosis; DIC: disseminated intravascular coagulation; EDTA: ethylenediaminetetraacetic acid; GPVI: glycoprotein VI; HRP: horseradish peroxidase; KCG: Κ-Carrageenan; LAT: linker activated T cells; LDS: lithium dodecyl sulfate; LTA: light-transmittance aggregometry; MFI: mean fluorescence intensity; PFA: paraformaldehyde; PLCγ2: phospholipase C gamma 2; PS: phosphatidylserine; Syk: spleen tyrosine kinase; Co-HP: Cobalt-hematoporphyrin
Introduction
In thrombosis research, animal models represent valuable tools to evaluate the effectiveness of pharmacotherapy and bridge the gap between in vitro studies and clinical applications.Citation1,Citation2 It is important to understand the pathogenic basis of thrombosis in small animals, which differs slightly from those in humans.
Kappa-carrageenan (KCG) is a water-soluble sulfated polysaccharide derived from a red algal cell wall.Citation3 It consists of alternating long linear chains of alpha-1,3 D-galactose and beta-3,6-anhydro-galactose with sulfate esters (15%—40%).Citation3 In the food industry, KCG is a well-researched ingredient that is frequently utilized as a thickener and stabilizer in several foods, owing to its gel-forming ability.Citation4,Citation5 In thrombosis research, Bekemeier et al. first reported that intraperitoneal administration of KCG induces tail thrombosis in mice or rats.Citation6,Citation7 They established the KCG-induced tail thrombosis (CTT) model as a valuable tool for preclinical studies of thrombosis and disseminated intravascular coagulation. The CTT model offers a simple technique without anesthesia and easy quantification to determine the necrotic region of the tail without euthanizing the laboratory animals. Previous studies reported that KCG induces biphasic aggregation in human platelet-rich plasma; aspirin and theophylline inhibit the second phase, while ethylenediaminetetraacetic acid (EDTA) inhibits both phases.Citation8 Another study by Hatmi et al. revealed that KCG-stimulated platelets produce intracellular inositol triphosphate 3 and diacylglycerol as secondary messengers, which are then inhibited by neomycin (used as a phospholipase C inhibitor).Citation9 While KCG-induced platelet activation has been reported in human and rabbit studies, the cell-surface receptor and essential signaling pathway remain unclear, as does its relationship with the CTT murine model.
In this study, we aimed to investigate the molecular basis of KCG-induced platelet activation and its relevance to CTT model. We attempted to examine the essential signaling pathways and pivotal cell-surface receptor of KCG using light-transmittance aggregometry (LTA), flow cytometry, and western blotting.
Methods
Kappa-Carrageenan (KCG)
KCG was purchased from Merck (Darmstadt, Germany, catalog number: 22048). It was dissolved in the normal saline at concentrations ranging from 6.25 µg/mL to 1000 µg/mL. For each experiment, we prepared the KCG solutions using the powder immediately before use.
Ethical considerations and animal use
The present study was conducted in accordance with the ethical principles and guidelines established by University of Yamanashi and was designed to ensure compliance with the ethical norms stipulated in the Declaration of Helsinki. Written informed consent was obtained from all healthy human donors before their inclusion in the study. The confidentiality and anonymity of the participants were strictly maintained throughout the research process.
Seven- to twelve-week-old male C57BL/6 mice obtained from CLEA Japan (Shizuoka, Japan) were used. Platelet-specific conditional C-type lectin-like receptor-2 (CLEC-2)-knock-out mice (Clec1bflox/flox PF4-Cre) and Clec1bflox/flox mice were generated as previously described.Citation10,Citation11 Male or female mice, aged were 8- to 14-weeks, were used.
The Animal Care and Use Committee at the University of Yamanashi approved this study (Experiment plan approval number: A5–12), and all animal experiments were conducted in accordance with their established guidelines.
Light transmittance aggregometry
We obtained murine blood through cardiac puncture after anesthetizing and euthanizing the mice using CO2. Washed murine platelets were then prepared following the previously described method,Citation12 using 10% acid citrate dextrose (ACD) and prostaglandin I2 (PGI2). The washed platelets were subsequently suspended in a modified Tyrode’s buffer, following the previously established protocol.Citation13 Platelet aggregation (200 × 103/µL) was determined using light transmittance aggregometry (LTA; MSM Hematracer 712, LMS, Tokyo, Japan). The washed murine platelets were then treated with various stimuli, including KCG at concentrations of 12.5–500 µg/mL, collagen-related peptide (CRP), α-thrombin, proteinase-activated receptor-4 agonist peptide (PAR-4 Ap), and recombinant rhodocytin.Citation14 To achieve the desired concentrations, inhibitors, including SU6656 (Src family kinase inhibitor), CAS-622387 (Syk inhibitor), YM-254890 (Gαq inhibitor), and TAK-242 (Toll-like receptor 4 inhibitor) were dissolved in dimethyl sulfoxide (DMSO). Prior to adding the stimuli, the inhibitors were pre-incubated for 5 min at 37°C. The concentrations of SU6656 and CAS-622387 were chosen for their efficacy in preventing the platelet aggregation with 0.2 µg/mL CRP, while the standard concentrations of YM-254890 was selected for its ability to inhibit 100 µM PAR-4 Ap-induced platelet aggregation.
To prepare human washed platelets, venous blood was drawn from healthy volunteers who were not taking any medications and mixed with 10% sodium citrate. By centrifuging the whole blood at 160 g for 10 min, platelet-rich plasma (PRP) was isolated. The platelets were subsequently rinsed twice in a buffer containing 15% acid-citrate dextrose and 1 µM PGI2. Finally, they were resuspended in modified Tyrode’s buffer to a concentration of 200 × 103/mL. Cobalt-hematoporphyrin (Co-HP) was prepared and used according to previous studies.Citation15 More information about reagents and antibodies used in this study can be found in Table S1.
Flow cytometry analysis
Platelet activation was evaluated using Accuri C6 Flow Cytometer (BD Biosciences), phycoerythrin (PE)-conjugated anti-mouse P-selectin (CD62P) antibody and PE-conjugated anti-activated integrin αIIbβ3 antibody (JON/A). Data analysis was performed using FlowJo 10.8.2 software. Washed murine platelet (100 × 103/µL) were incubated with a PE-conjugated rat IgG1 Isotype control and CD62P or JON/A antibodies for 1 h after stimulation. The final concentration of the stimulation was the same as that used in the LTA assay. The reaction was inhibited with 0.1% paraformaldehyde (PFA) and analyzed by using flow cytometry. Platelets were rated based on size, and 10 000 single platelet events were counted to compare the mean fluorescence intensity (MFI). In the Annexin V-binding assay, washed murine platelets (500 × 103/µL) were treated with a stimulant under static conditions at 37°C for 30 min and then resuspended in Annexin binding buffer (1:5). Annexin V-FITC (5 µL) was then added to the stimulated platelets, and the mixture was incubated for an additional 30 min. Flow cytometry measured the FITC signal immediately during the Annexin V-FITC reaction. Annexin V-FITC-positive platelets were calculated as the percentage of positive platelets in the stimulated group, with a cutoff of 2% for no stimulation.
Western blotting
The washed murine platelets were stimulated in HemaTracer® 712. Stimulation was applied at a concentration of 1 mg/mL of KCG and was stopped with a lithium dodecyl sulfate (LDS) sample buffer. Control stimulation was performed using saline, CRP, and rhodocytin. The inhibitors were incubated with 100 µM CAS-622387 and 2 µM YM-254890 for 10 min prior to adding the agonists. The lysates were denatured at 70°C for 15 min, separated using NuPAGE 4%–12% Bis-Tris Protein Gels and electrophoresed in MOPS SDS running buffer. A BlueStar protein marker was used as a reference. Proteins were then transferred to a membrane using the Trans-Blot Turbo Transfer Kit following the manufacturer’s instructions. The membranes were blocked with Blocking One-P and incubated with primary antibodies (phosphotyrosine antibody [4G10]), spleen tyrosine kinase (Syk) antibody, phospho-Syk antibody (Tyr525/526), phospholipase Cγ2 (PLCγ2) antibody), phosphor-PLCγ2 antibody (Tyr1217), linker for activation of T cells (LAT) antibody, phospho-LAT antibody (Tyr191), and anti-β-actin antibody at 1 µg/mL in Signal Enhancer HIKARI for 12 h at room temperature (average 18–24°C). After washing, the samples were exposed to secondary antibodies conjugated with horseradish peroxidase (HRP). The protein bands were detected using the ECL Prime enhanced chemiluminescence reagent obtained from Cytiva. The ImageQuantTM LAS4000 mini captured the protein bands.
Surface plasmon resonance (SPR) spectroscopy analysis
The direct binding of KCG to recombinant CLEC-2 was assessed using Biacore X (Biacore AB, Uppsala, Sweden). Recombinant human or mouse CLEC-2 chimera and recombinant mouse podoplanin were prepared according to methods outlined in previous studies.Citation16 Human CLEC-2-rabbit Fc, mouse CLEC-2-rabbit Fc, and rabbit Fc (used as a control) were covalently attached to CM5 chips (GE Healthcare) using the amine coupling kit and were then solid-phased. Various concentrations of KCG, diluted in saline, were perfused onto the recombinant coat surface at a rate of 20 μL/min at 25°C, and the resulting changes in resonance units (RU) were recorded. NaOH was used as the regeneration solution and injected into the system to cleave the specific binding of the analyte to the ligand. The RU for the KCG/CLEC-2 interaction was calculated by subtracting the RU of rabbit Fc from the RU of either human CLEC-2-rabbit Fc or mouse CLEC-2-rabbit Fc. To create a positive control and to ensure that the recombinant CLEC-2 was properly immobilized, the binding of CLEC-2 to rhodocytin or podoplanin was confirmed.
Assessment of KCG-induced tail thrombosis in mice
KCG (80 µg/g body weight) was administered intraperitoneally, and the tail thrombus was evaluated 24 h after the tail skin was removed following CO2- assisted euthanasia. The venous thrombus rate (%) was assessed as the length of the venous thrombus divided by the total tail length. Murine blood was collected with 10 mM EDTA by cardiac puncture, and platelet count was measured using a blood cell analyzer XE-5000 (Sysmex, Hyogo, Japan). The mice were then fixed via intracardiac perfusion fixation with 4% PFA to prepare the tissue sections. Samples were prepared using hematoxylin and eosin (H&E) staining after decalcification with K-CX for 24 h and paraffin embedding, taken at 3 cm from the tail tip.
Statistical analysis
The data were subjected to statistical analysis using various methods, including unpaired Student’s t-test, two-way ANOVA with the Bonferroni test, or one-way ANOVA with Dunnett’s multiple comparison tests in GraphPad Prism 9 (GraphPad Software, Inc., San Diego, CA, US). The results are reported as the mean ± standard deviation.
Results
KCG induces platelet activation in mice
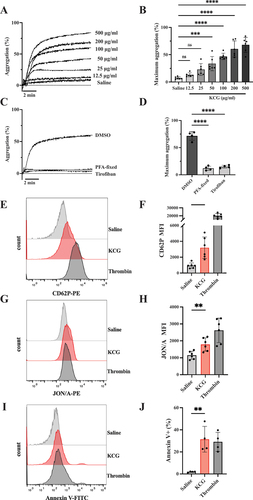
We first confirmed murine platelet aggregation by KCG in LTA assay. KCG induced an initial shape change and dose-dependent platelet aggregation (). Notable increase in platelet aggregation was observed when more than 50 μg/mL of KCG was used compared to the saline (). We observed that 100 µg/mL of KCG induced stably reproducible platelet aggregation in mice; therefore, we utilized KCG at this concentration for further experimentation. KCG-induced platelet aggregation was abolished in PFA-fixed platelets () and pretreatment with tirofiban, an inhibitor of integrin αIIbβ3, also completely inhibited KCG-induced platelet aggregation (). These results suggest KCG-induced platelet aggregation is mediated by platelet-platelet adhesion with activated integrin αIIbβ3 rather than by agglutination. We then evaluated p-selectin (CD62P) expression as an α-granule secretion and JON/A binding as activation of integrin αIIbβ3. KCG-stimulated platelets showed p-selectin expression and activated integrin αIIβ3 compared to saline-stimulated platelets (). We also examined Annexin V binding to evaluate phosphatidylserine (PS) exposure. KCG-exposed platelets significantly increased PS exposure comparable to thrombin-stimulated platelets (). These data suggest that KCG causes murine platelet activation leading to α-granules secretion, activated integrin αIIbβ3, and PS exposure on the platelet surface.
Figure 1. Kappa-carrageenan (KCG) induces platelet activation in mice. (A) Washed murine platelets (200×103/µL) were stimulated with the indicated concentrations of KCG for 10 min using the LTA assay. Representative aggregation curves are shown. (B) Quantified maximum aggregation (%) (n = 8). Statistical significance was analyzed by one-way ANOVA with Dunnett’s multiple comparison test; ***P < .001, ****P < .0001, and not significant (ns). (C) Platelet aggregation stimulated with KCG (100 µg/mL) was evaluated in platelets once PFA-fixed or, pretreated with DMSO or tirofiban. The platelets were pre-incubated with 1% DMSO or 0.5 µM tirofiban for 5 min before stimulation. Murine platelets were fixed once with 0.1% PFA, and PFA was washed off, resuspended in modified Tyrode’s buffer to 200×103/µL and stimulated with KCG. (D) Quantified maximum aggregation (%) (n = 4). Statistical significance was analyzed by one-way ANOVA with Dunnett’s multiple comparison tests, where ****P < .0001. (E) Washed murine platelets (100×103/µL) were stimulated with saline, KCG (100 µg/mL), and thrombin (0.1 U/mL) followed by incubation with the anti-CD62P antibody for 1 h. The reactions were terminated with 0.1% PFA and analyzed by using flow cytometer. Representative histograms (n = 6) were created using the FlowJo software. (F) Quantified mean fluorescent intensity (MFI) of CD62P (n = 6). (G) The protocol was the same as (E), except the JON/A antibody was used. Representative histograms of JON/A binding are shown. (H) Quantified MFI of JON/A (n = 6). (I) Washed murine platelets (500×103/µL) were treated with saline, KCG (100 µg/mL), and thrombin (0.25 U/mL) for 30 min and then resuspended in Annexin binding buffer. After additional incubation with Annexin V-FITC, the FITC signal was measured by using flow cytometry immediately. Representative histograms of Annexin V binding are shown. (J) Summary data showing the percentage of Annexin V positive platelets (%) are presented (n = 4). Statistical significance was assessed by using a two-tailed, unpaired Student’s t-test; **P < .001.
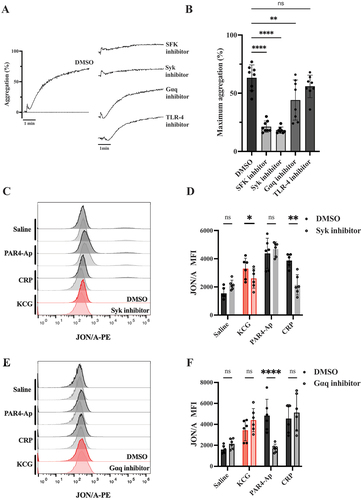
Src family kinases (SFK) and Syk are required for KCG-induced platelet activation
After observing the platelet activation with KCG, we attempted to identify the critical pathways of platelet activation by testing inhibitors. ( shows that pretreatment with SU6656 (SFK inhibitor) and CAS-622387 (Syk inhibitor) significantly inhibited KCG-induced aggregation, indicating that SFK and Syk are predominantly responsible for KCG-induced aggregation. Pretreatment with YM-254890 (Gαq inhibitor) led to a partial reduction, while the G protein-coupled receptor (GPCR) pathways are partially involved, possibly through feedback from thromboxane A2. The concentrations of SFK and Syk inhibitors used in this experiment were determined based on their ability to inhibit platelet aggregation induced by 0.2 µg/mL CRP, an agonist of glycoprotein VI, which is highly dependent on SFK and Syk. The concentration of Gαq inhibitor was determined as the concentration that inhibits platelet aggregation induced by 100 µM PAR4-Ap, because PAR-4 couples Gαq (Figure S1). We also tested Toll-like receptor 4 (TLR-4) inhibitors TAK-242, as previous studies have shown that KCG activates macrophages via TLR-4.Citation17 TLR-4 is also expressed in plateletsCitation18 and its ligand, lipopolysaccharide (LPS), induces platelet activation, a matter that is debatable.Citation19 The concentration of TAK-242 utilized in this study was determined based on its ability to inhibit LPS-induced murine platelet aggregation (Figure S2A–B). TAK-242 failed to inhibit KCG-induced platelet aggregation (). We then evaluated JON/A binding using CAS-622387 and YM-254890 to further support the Syk-dependent pathway, not the GPCR pathway in KCG-induced platelet activation. The integrin αIIβ3 activation caused by KCG was significantly reduced in platelets pretreated with CAS-622387 but not reduced when using YM-254890 (). These results demonstrate that the SFK- and Syk-dependent pathways are essential for KCG-mediated platelet activation.
Figure 2. KCG activates murine platelets in a tyrosine-kinase-dependent manner (A) Washed murine platelets (200 × 103/µL) were pretreated with SFK inhibitor (SU6656) (20 µM) Syk inhibitor (CAS-622387) (20 µM), Gαq inhibitor (YM-254890) (0.4 µM), and TLR-4 inhibitor (TAK-242) (10 µM) for 5 min and then stimulated with KCG (100 µg/mL). All inhibitors were diluted in DMSO, and controls were tested with final DMSO concentrations ranging from 0.1–2%. Representative aggregation curves are shown. (B) Maximum aggregation data were quantified (n = 8), and the statistical significance was analyzed by using one-way ANOVA with Dunnett’s multiple comparison tests; ***P < .01, ****P < .0001, and not significant (ns). (C) Platelets were pre-incubated with 0.5% DMSO or Syk inhibitor (CAS-622387) (20 µM) for 5 min and then stimulated with KCG (100 µg/mL), PAR4-Ap (100 µM), and CRP (0.2 µg/mL). After incubation for 1 h at 37°C, JON/A binding was measured by using flow cytometry. Representative histograms are shown. (D) Mean fluorescence intensity (MFI) of JON/A was quantified (n = 6). (E) Platelets were pre-incubated with 0.2% DMSO or Gαq inhibitor (YM-254890) (0.4 µM) for 5 min and then stimulated with agonists for 1 h. Representative histograms are shown. (F) MFI of JON/A was quantified (n = 6). Data were statistically analyzed by using two-way ANOVA with the Dunnett’s test; ****P < .05; **P < .01; **P < .0001, and not significant (ns).
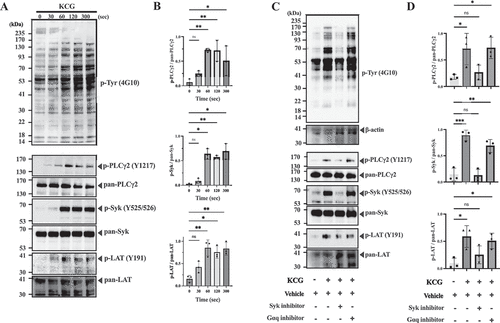
KCG induced intracellular tyrosine phosphorylation in a time-dependent manner
We next evaluated intracellular signaling mediated by KCG. According to the tyrosine kinase-dependent pathway required for KCG-induced platelet aggregation, we examined the pan-tyrosine phosphorylation and tyrosine phosphorylation of Syk, LAT, and PLCγ2 (). Platelets exposed to KCG exhibited a time-dependent increase of pan-tyrosine phosphorylation in whole cell lysates. Further, they showed tyrosine phosphorylation of the Syk, LAT, and PLCγ2 within a minute as well as those stimulated with CRP and rhodocytin. We compared the intracellular tyrosine phosphorylation patterns during KCG-induced platelet activation with those observed during CRP- or rhodocytin-induced platelet activation. Platelets exposed to KCG exhibited attenuated phosphorylation of total protein (Figure S3A), Syk, PLCγ2, and LAT (Figure S3A–B) compared to when they were exposed to CRP and rhodocytin. Tyrosine phosphorylation was significantly inhibited by pretreatment with CAS-622387 but not with YM-254890 (). These data emphasize that KCG-mediated platelet activation is a tyrosine kinase-dependent pathway.
Figure 3. KCG induces intracellular tyrosine phosphorylation (A) Washed murine platelets (1000 × 103/µL) were stimulated with KCG (500 µg/mL), and the platelet reaction was stopped at the indicated second. The whole cell lysates were subjected to sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting with antibodies against anti-phosphotyrosine antibody (4G10), phosphorylated-Syk(Y526/526), total-Syk, phosphorylated-PLC (Y1217), total-PLCγ2, phosphorylated-LAT(Y191), and total-LAT. (B) This experiment was performed three times independently, and the signal intensity of specific phosphorylated tyrosine proteins was quantified using ImageJ software. One-way ANOVA statistically analyzed data (n = 3) with the Dunnett’s test; ***P < .05 with ****P < .01 and not significant (ns) (C) Washed murine platelets (1000 × 103/µL) were pre-incubated with Syk inhibitor (CAS-622387) (100 µM) or Gαq inhibitor (YM-254890) (2 µM) for 10 min. After KCG stimulation (500 µg/mL), the platelet reaction was terminated at 60 sec with LDS sample buffer. SDS-PAGE and western blotting conditions were performed as described above. The images are representative of three independent experiments. (D) The results of three experiments were quantified and statistically analyzed with one- way ANOVA with Dunnett’s tests; ****P < .01; **P < .001, and not significant (ns).
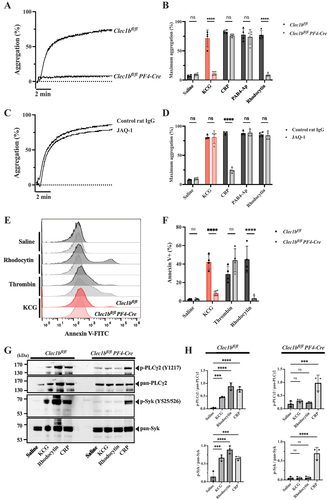
CLEC-2 is the essential receptor for KCG-induced platelet aggregation
Murine platelets possess two important receptors for tyrosine kinase-mediated activation: GPVI-FcRγ and CLEC-2.Citation20 GPVI-FcRγ serves as a receptor for collagen, whereas platelet CLEC-2 acts as a receptor for rhodocytin/podoplanin. To identify the essential platelet receptor of KCG, we examined platelets from platelet/megakaryocyte-specific CLEC-2-deficient mice (Clec1bfl/fl Pf4-Cre mice) and platelets pretreated with anti-GPVI blocking antibody, JAQ1. Similar to rhodocytin-induced aggregation, KCG-induced platelet aggregation was completely abolished in CLEC-2-deficient platelets (). JAQ1 blocked CRP-induced platelet aggregation but failed to inhibit KCG-induced platelet aggregation (). We also confirmed that PS exposure induced by KCG was significantly prevented in CLEC-2-deficient platelets compared to wild-type platelets as caused by rhodocytin (). Furthermore, the KCG-mediated intracellular phosphorylation of Syk and PLCγ2 was significantly suppressed in CLEC-2-deficient platelets compared to wild-type platelets ().
Figure 4. CLEC-2 is the essential receptor in KCG-induced platelet activation (A) Representative platelet aggregation curves are shown for platelets from Clec1bfl/fl mice and Clec1bfl/fl PF4-Cre mice, followed by KCG stimulation (100 µg/mL). (B) Quantified maximum aggregation stimulated with saline, KCG, CRP (0.2 µg/mL), PAR4-Ap (100 µM), and rhodocytin (10 nM) was quantified (n = 4). (C) Representative platelet aggregation curves are shown for murine platelets pre-incubated with control rat IgG or JAQ1 antibody (20 µM) for 10 min, followed by KCG (100 µg/mL) stimulation. (D) Quantified maximum aggregation stimulated with saline, KCG, CRP (0.2 µg/mL), PAR4-Ap (100 µM), and rhodocytin (10 nM) was quantified (n = 4). (E) Representative histograms showing Annexin-V FITC positive platelets are presented for murine platelets obtained from Clec1bfl/fl mice and Clec1bfl/fl Pf4-Cre mice. Platelet were stimulated with saline, rhodocytin (100 nM), thrombin (0.5 U/mL), and KCG (500 µg/mL) for 1 h at 37°C. (F) Quantitative information (n = 4). Statistical analysis was performed using two-way ANOVA with the Bonferroni test, and statistical significance is defined as ***P < .05, ****P < .01, ****P < .001, **P < .0001, and not significant (ns). (G) Washed platelets (500 × 103/µL) from Clec1bfl/fl mice and Clec1bfl/fl Pf4-Cre mice were stimulated with KCG (500 µg/mL), rhodocytin (100 nM), or CRP (2.0 µg/mL). The reaction was stopped after 60 sec, and platelet lysates were collected. The samples were subjected to western blotting using the indicated antibodies on the same membrane. A representative example of the data obtained from three independent experiments is shown. (H) Quantitative information from the three independent experiments and statistically analyzed with one-way ANOVA with Dunnett’s comparison tests, where **P < .001 and not significant (ns).
KCG directly binds to recombinant mouse CLEC-2 and activates human platelets in a CLEC-2-dependent manner
We confirmed the direct binding of KCG to CLEC-2 using SPR spectroscopy. Recombinant mouse CLEC-2-rabbit Fc was prepared as previously described,Citation16 and KCG, at various dilutions, was used for the analytes with the Biacore sensor system. The results demonstrated that KCG binds to mCLEC-2 (). Rhodocytin and podoplanin are widely recognized ligands for CLEC-2. The binding of these ligands to CLEC-2,, are also shown in the results (). The binding affinity of KCG to CLEC-2 was weaker than that of rhodocytin or podoplanin to CLEC-2.
Figure 5. SPR analysis of KCG binding to mouse CLEC-2 (A) SPR data: sensorgrams were generated using a Biacore X system. Recombinant mouse CLEC-2-rFc and rabbit-Fc were immobilized via amine coupling on a CM5 sensor chip. KCG, diluted from 50 µg/mL to 1000 µg/mL in saline, served as the analyte in the solution phase. The resonance unit (RU) was plotted with rabbit-Fc as the reference, illustrating the binding to CLEC-2-rFc. (B) Rhodocytin, diluted from 25 µg/mL to 200 µg/mL, was used as an analyte to assess its binding RU to mCLEC-2 on the same sensor chip. (C) Mouse recombinant podoplanin, diluted from 25 µg/mL to 500 µg/mL, was evaluated as an analyte.
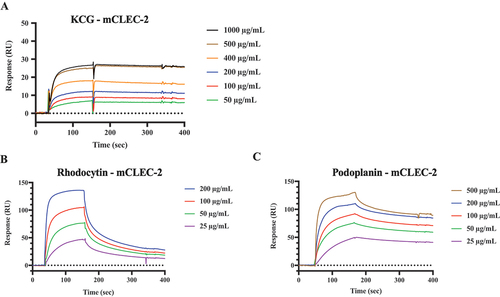
We also investigated whether KCG activates platelets via CLEC-2 in humans. KCG induced human platelet aggregation in a dose-dependent manner; aggregation induction increased considerably at KCG concentrations above 50 µg/mL (Figure S4A,B). A chemical inhibitor, Co-HP, which we have reported previously,Citation15 noticeably inhibited KCG-induced platelet aggregation. Additionally, KCG bound to recombinant human CLEC-2, as well as rhodocytin; nevertheless, the binding intensity of KCG to CLEC-2 was weaker than that of KCG to rhodocytin ().
Our in vitro results indicated that platelet surface receptor CLEC-2 and its downstream tyrosine kinase-dependent activation is the molecular basis of KCG-induced platelet aggregation in mice and that KCG also induces platelet aggregation in a CLEC-2 dependent manner in humans.
CLEC-2 -deficient mice were significantly inhibited in KCG-induced tail thrombosis
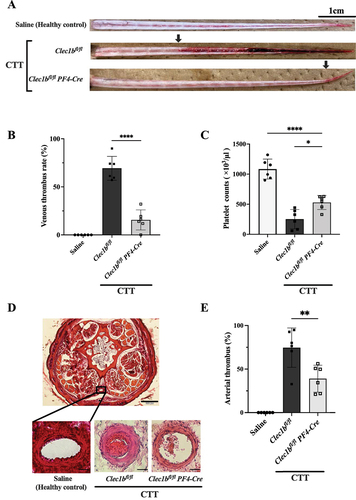
We finally evaluated the role of platelet CLEC-2 in vivo. We established the CTT murine model in wild-type mice (Clec1bfl/fl mice) and CLEC-2-deficient mice (Clec1bfl/fl PF4-Cre mice). We then evaluated the venous thrombus rate and thrombocytopenia 24 h after the intraperitoneal injection of KCG. The saline injection group was used as a healthy control. The method to measure the venous thrombosis rate, which involves observing the tails after skin removal, is illustrated in Figure S5A. As shown in (, the venous thrombus rate was 69.27 ± 12.44% (mean ± SD) in the wild-type mice. In contrast, in CLEC-2-deficient mice, the rate was significantly lower at 15.59 ± 10.42% (mean ± SD), indicating that the absence of platelet CLEC-2 significantly inhibited the CTT murine model. Further, thrombocytopenia was significantly (*P < .05) but partially rescued in the CLEC-2-deficient mice (); however, Clec1bfl/fl Pf4-Cre mice have been reported to exhibit thrombocytopenia.Citation21 As KCG has been reported to induce both arterial and venous thrombosis in mice, we also evaluated the microthrombus area of the caudal artery. The section of arterial microthrombus formation 3 cm away from the tail tip also showed a marked reduction in microthrombus in CLEC-2-deficient mice (). Moreover, when we evaluated thrombosis in additional organs, we notably observed substantial thrombus formation in the hepatic portal vein. This suggests that KCG administered intraperitoneally may be partially absorbed via the omental veins, leading to rapid clotting in the portal veins (Figure S5B). Importantly, the incidence of portal vein thrombosis was considerably lower in CLEC-2-deficient mice (Figure S5C).
Figure 6. Platelet CLEC-2 deficiency inhibits KCG-induced tail thrombosis (A) Representative images of macrothrombus in the caudal vein 24 h after KCG administration, with the tail skin removed, are shown for healthy control, Clec1bfl/fl mice and Clec1bfl/fl Pf4-Cre mice. The arrowhead indicates the borders of the venous thrombus, scale bar = 1 cm. (B) quantification data of venous thrombus rate (%) with three groups (n = 6). Statistical significance was assessed by using a two-tailed, unpaired Student’s t-test; ***P < .0001. (C) thrombocytopenia was observed in the CTT groups 24 h after KCG administration. Data were analyzed by using one-way ANOVA with Dunnett’s multiple comparison tests; ****P < .05 and ****P < .0001. (D) microthrombus formation in the caudal artery was evaluated. Representative hematoxylin and eosin (HE)-stained tail tissues are shown for a section taken 3 cm from the tail. In the magnified view of healthy control mice, Clec1bfl/fl mice, and Clec1bfl/fl PF4-Cre mice. The upper magnified view scale bar represents 200 µm, while the lower arterial magnified view scale bar indicates 20 µm. (E) ImageJ software analyzed the tissue sections’ vessel lumen and thrombus area. The arterial thrombus filling ratio was quantified (n = 6). Statistical significance was assessed by using a two-tailed, unpaired Student’s t-test; **P < .01.
Thus, our findings suggest that the CLEC-2-mediated prothrombotic state after KCG injection is substantially related to the CTT mouse model.
Discussion
In this study, we set out to elucidate the molecular basis of KCG-induced platelet activation within the CTT model, which has been used in preclinical studies. We showed that KCG triggers platelet aggregation as well as α-granule secretion, activated integrin αIIbβ3, and PS exposure on the surface of murine platelets (). KCG-exposed platelets exhibited intracellular tyrosine phosphorylation of Syk, LAT, and PLCγ2, and SFK- and Syk-dependent pathways are required for KCG-induced platelet aggregation (). CLEC-2-deficient platelets showed significant inhibition in KCG-induced platelet aggregation, indicating that CLEC-2 is an essential molecule in that reaction (). The SPR data () suggest the direct binding of KCG with CLEC-2. We then assessed the CTT murine model using Clec1bfl/fl Pf4-Cre mice and showed that venous thrombosis and arterial microthrombi were significantly inhibited in the CLEC-2-deficient mice (). These findings suggest that CLEC-2-mediated platelet activation leading to a prothrombotic state is partially the cause of the pathogenesis of the CTT model.
The CTT model developed by H. Bekemeier offers a noninvasive, high-frequency, and easily quantifiable option to screen antithrombotic drugs in systemic thrombosis.Citation22,Citation23 However, it has yet to become a standard model in thrombosis research, probably owing to the limited research on the molecular mechanisms of thrombosis. Antiplatelet drugs and herbal medicines have shown effectiveness against CTT,Citation24–26 but no study has pointed out that CLEC-2-mediated platelet activation is involved in CTT pathogenesis. Notably, KCG strongly induced PS-positive procoagulant platelets, as does those stimulated with thrombin (). Recent studies have also highlighted the subpopulation of procoagulant platelets with distinct properties.Citation27,Citation28 While we cannot definitively assert that CLEC-2-mediated signaling specifically promotes PS exposure, we found that KCG-induced platelet aggregation and annexin V binding were significantly attenuated in CLEC-2-deficient platelets (Figure S4E–F). KCG has been widely studied as a pro-inflammatory substance in preclinical studies to induce subcutaneous edema and colitis in experimental animals.Citation29–31 Recent studies show that KCG administration activates intraperitoneal macrophages via the TLR-4/CD14/MyD88 signaling pathway,Citation32 leading to pro-inflammatory cytokine production and endothelial cell injury, ultimately promoting the coagulation pathway.Citation33,Citation34 In our in vivo study, TAK-242 partially inhibited CTT thrombosis (Figure S5D). We attribute this to inhibition of the macrophage TLR-4-mediated intraperitoneal inflammation by TAK-242. Inflammation-triggered vascular endothelial damage and the form of neutrophil extracellular traps may be a part of the pathogenesis of this thrombosis model as previously reported.Citation34 Thus, the extrinsic coagulation pathway stimulated by intraperitoneal inflammation via macrophages has been suggested to be involved in CTT; however, the procoagulant state stimulated with KCG via platelet activation receptor CLEC-2, might be involved in the CTT model in large part.
Our study also suggests that in the case of sulfated polysaccharides, KCG acts as an exogenous ligand for CLEC-2, as previously reported for dextran sulfate and fucoidan,Citation35–37 which are different components of sulfated polysaccharides. Intracellular tyrosine phosphorylation signaling by KCG tended to be weaker than signaling by rhodocytin (Figure S3) even though we used the supra-maximal concentration of KCG as shown in the aggregation assay (). The molecular weight of rhodocytin (~120 kDa) is much smaller than that of KCG (~700 kDa),Citation38 but there are four CLEC-2 binding sites in one rhodocytin molecule.Citation14 The ability of rhodocytin to bind and cluster CLEC-2 May be greater than that of KCG, allowing rhodocytin to induce substantially more tyrosine phosphorylation.Citation39 The C-type lectin superfamily, to which platelet CLEC-2 belongs, has a wide range of physiological functions and recognizes both endogenous and exogenous ligands.Citation40 Thus, we posit that platelet CLEC-2 recognizes sulfated polysaccharides in some foreign antigens and functions as a receptor for pathogen-associated molecular patterns and CLEC-2-mediated platelet activation may participate in innate immunity via unknown mechanisms. In vivo, CLEC-2 May interact with internal sulfated polysaccharides in pathological conditions to induce some pathological effects. However, further research is required to explore this topic.
Our in vitro experiment results also showed that KCG induces human platelet aggregation (Figure S4), consistent with previous studies.Citation9 Furthermore, we demonstrated that platelet aggregation induced by KCG or rhodocytin, but not that induced by CRP or thrombin, was appreciably inhibited by Co-HP, a chemical inhibitor of CLEC-2 (Figure S4C–D). Using SRP assays, we also showed that recombinant human CLEC-2 directly binds to KCG; nevertheless, the binding intensity of CLEC-2 binding to KCG was weaker than that of rhodocytin binding to KCG (Figure S4E–F). These findings suggest that KCG induces platelet aggregation by binding to CLEC-2 not only in mice, but also in humans. Carrageenan (CG) is a natural polysaccharide extracted from red algae, commonly used as a thickening and gelling agent in foods and cosmetics. Global organizations, such as the World Health Organization and the Joint FAO Expert Committee on Food Additives, consider that the use of CG as a food additive is safe.Citation41 Functioning as a dietary fiber, CG is minimally digested in the intestine, making it unlikely to enter the bloodstream in humans. Therefore, we assume that once CG does enter bloodstream, it induces platelet aggregation by binding to CLEC-2 in humans as in mice; however, this does not happen under normal circumstances.
In conclusion, our findings indicate that KCG induces platelet aggregation via CLEC-2, and the CLEC-2-mediated prothrombotic state is involved in the CTT murine model. As the CTT murine model is used to screen antithrombotic drugs, our new understanding of the molecular basis of CTT may contribute to developing new thrombotic disease treatments.
Supplemental Material
Download PDF (1.1 MB)Acknowledgments
We would like to express our sincere appreciation to Takako Otsuka and Syouko Saigusa for their invaluable support in conducting the experiments and analyzing the data. We would also like to acknowledge the contributions of Naoki Kosaka in data collection and laboratory work. Additionally, we extend our thanks to Editage (www.editage.com) for their assistance with English language editing.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data necessary to understand and assess the findings of this study are provided within the main text and Supplementary Figures.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/09537104.2023.2281941.
Additional information
Funding
References
- Diaz JA, Obi AT, Myers DD Jr., Wrobleski SK, Henke PK, Mackman N, Wakefield TW. Critical review of mouse models of venous thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:556–11. doi:10.1161/atvbaha.111.244608.
- Albadawi H, Witting AA, Pershad Y, Wallace A, Fleck AR, Hoang P, Khademhosseini A, Oklu R. Animal models of venous thrombosis. Cardiovasc Diagn Ther. 2017;7(S3):S197–s206. doi:10.21037/cdt.2017.08.10.
- Jiao G, Yu G, Zhang J, Ewart HS. Chemical structures and bioactivities of sulfated polysaccharides from Marine algae. Mar Drugs. 2011;9(2):196–223. doi:10.3390/md9020196.
- Necas J, Bartosikova L. Carrageenan: a review. Vet Med (Praha). 2013;58(4):187–205. doi:10.17221/6758-VETMED.
- Yegappan R, Selvaprithiviraj V, Amirthalingam S, Jayakumar R. Carrageenan based hydrogels for drug delivery, tissue engineering and wound healing. Carbohyd Polym. 2018;198:385–400. doi:10.1016/j.carbpol.2018.06.086.
- Bekemeier H, Hirschelmann R, Giessler AJ. Carrageenin-induced thrombosis in the rat and mouse as a test model of substances influencing thrombosis. Biomed Biochim Acta. 1984;43:S347–350.
- Bekemeier H, Hirschelmann R, Giessler AJ. Carrageenin-induced thrombosis in rats and mice: a model for testing antithrombotic substances? Agents Actions. 1985;16:446–51. doi:10.1007/bf01982887.
- Kindness G, Long WF, Williamson FB, Boyd J. Effects of carrageenans on the aggregation of human blood platelets. Thromb Res. 1979;15:3–15. doi:10.1016/0049-3848(79)90047-1.
- Hatmi M, Randon J, Faili A, Vargaftig BB. Carrageenan-induced activation of human platelets is dependent on the phospholipase C pathway. Br J Haematol. 1993;83:270–5. doi:10.1111/j.1365-2141.1993.tb08282.x.
- Suzuki-Inoue K, Inoue O, Ding G, Nishimura S, Hokamura K, Eto K, Kashiwagi H, Tomiyama Y, Yatomi Y, Umemura K, et al. Essential in vivo roles of the C-type lectin receptor CLEC-2: embryonic/neonatal lethality of CLEC-2-deficient mice by blood/lymphatic misconnections and impaired thrombus formation of CLEC-2-deficient platelets. J Biol Chem. 2010;285:24494–507. doi:10.1074/jbc.M110.130575. Epub 2010/06/08.
- Osada M, Inoue O, Ding G, Shirai T, Ichise H, Hirayama K, Takano K, Yatomi Y, Hirashima M, Fujii H, et al. Platelet activation receptor CLEC-2 regulates blood/lymphatic vessel separation by inhibiting proliferation, migration, and tube formation of lymphatic endothelial cells. J Biol Chem. 2012;287:22241–52. doi:10.1074/jbc.M111.329987. Epub 2012/05/05.
- Inoue O, Hokamura K, Shirai T, Osada M, Tsukiji N, Hatakeyama K, Umemura K, Asada Y, Suzuki-Inoue K, Ozaki Y. Vascular smooth muscle cells stimulate platelets and facilitate thrombus formation through platelet CLEC-2: Implications in Atherothrombosis. PloS One. 2015;10:e0139357. doi:10.1371/journal.pone.0139357. Epub 20150929.
- Suzuki-Inoue K, Inoue O, Frampton J, Watson SP. Murine GPVI stimulates weak integrin activation in PLCgamma2-/- platelets: involvement of PLCgamma1 and PI3-kinase. Blood. 2003;102:1367–73. doi:10.1182/blood-2003-01-0029. Epub 20030501.
- Sasaki T, Shirai T, Tsukiji N, Otake S, Tamura S, Ichikawa J, Osada M, Satoh K, Ozaki Y, Suzuki-Inoue K. Functional characterization of recombinant snake venom rhodocytin: rhodocytin mutant blocks CLEC-2/podoplanin-dependent platelet aggregation and lung metastasis. J Thromb Haemost. 2018;16:960–72. doi:10.1111/jth.13987. Epub 20180330.
- Tsukiji N, Osada M, Sasaki T, Shirai T, Satoh K, Inoue O, Umetani N, Mochizuki C, Saito T, Kojima S, et al. Cobalt hematoporphyrin inhibits CLEC-2-podoplanin interaction, tumor metastasis, and arterial/venous thrombosis in mice. Blood Adv. 2018;2:2214–25. doi:10.1182/bloodadvances.2018016261.
- Suzuki-Inoue K, Kato Y, Inoue O, Kaneko MK, Mishima K, Yatomi Y, Yamazaki Y, Narimatsu H, Ozaki Y. Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J Biol Chem. 2007;282:25993–6001. doi:10.1074/jbc.M702327200. Epub 2007/07/10.
- Myers MJ, Deaver CM, Lewandowski AJ. Molecular mechanism of action responsible for carrageenan-induced inflammatory response. Mol Immunol. 2019;109:38–42. doi:10.1016/j.molimm.2019.02.020. Epub 20190307.
- Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, Ni H, Lazarus AH, Freedman J, Semple JW. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. 2006;107:637–41. doi:10.1182/blood-2005-06-2202. Epub 20050922.
- Galgano L, Guidetti GF, Torti M, Canobbio I. The controversial role of LPS in platelet activation in vitro. Int J Mol Sci. 2022;23: doi:10.3390/ijms231810900. Epub 20220917.
- Rayes J, Watson SP, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest. 2019;129:12–23. doi:10.1172/jci122955. Epub 20190102.
- Tamura S, Suzuki-Inoue K, Tsukiji N, Shirai T, Sasaki T, Osada M, Satoh K, Ozaki Y. Podoplanin-positive periarteriolar stromal cells promote megakaryocyte growth and proplatelet formation in mice by CLEC-2. Blood. 2016;127:1701–10. doi:10.1182/blood-2015-08-663708. Epub 20160121.
- Bekemeier H, Giessler AJ. Thrombosis induction by different carrageenans in rats and mice. Naturwissenschaften. 1987;74:345–6. doi:10.1007/bf00367934.
- Bekemeier H, Hirschelmann R. Role of eicosanoids in the kappa-carrageenin rat tail thrombosis. Biomed Biochim Acta. 1988;47:S260–263.
- Hirschelmann R, Bekemeier H. Heparin and phenprocoumon inhibit kappa-carrageenin induced thrombosis in rats. Folia Haematol Int Mag Klin Morphol Blutforsch. 1986;113:379–82.
- Ma N, Liu XW, Yang YJ, Li JY, Mohamed I, Liu GR, Zhang JY. Preventive effect of aspirin eugenol ester on thrombosis in κ-carrageenan-induced rat tail thrombosis model. PloS One. 2015;10:e0133125. doi:10.1371/journal.pone.0133125. Epub 20150720.
- Zhang B, Huang R, Yang D, Chen G, Chen Y, Han J, Zhang S, Ma L, Yang X. Combination of colchicine and ticagrelor inhibits carrageenan-induced thrombi in mice. Oxid Med Cell Longev. 2022;2022:3087198. doi:10.1155/2022/3087198. Epub 20220117.
- Topalov NN, Yakimenko AO, Canault M, Artemenko EO, Zakharova NV, Abaeva AA, Loosveld M, Ataullakhanov FI, Nurden AT, Alessi MC, et al. Two types of procoagulant platelets are formed upon physiological activation and are controlled by integrin α(IIb)β(3). Arterioscler Thromb Vasc Biol. 2012;32:2475–83. doi:10.1161/atvbaha.112.253765. Epub 20120726.
- Agbani EO, Poole AW. Procoagulant platelets: generation, function, and therapeutic targeting in thrombosis. Blood. 2017;130:2171–9. doi:10.1182/blood-2017-05-787259. Epub 20170927.
- Morris CJ. Carrageenan-induced paw edema in the rat and mouse. Methods Mol Biol. 2003;225:115–21. doi:10.1385/1-59259-374-7:115.
- Shang Q, Sun W, Shan X, Jiang H, Cai C, Hao J, Li G, Yu G. Carrageenan-induced colitis is associated with decreased population of anti-inflammatory bacterium, akkermansia muciniphila, in the gut microbiota of C57BL/6J mice. Toxicol Lett. 2017;279:87–95. doi:10.1016/j.toxlet.2017.07.904. Epub 20170801.
- Mi Y, Chin YX, Cao WX, Chang YG, Lim PE, Xue CH, Tang QJ. Native κ-carrageenan induced-colitis is related to host intestinal microecology. Int J Biol Macromol. 2020;147:284–94. doi:10.1016/j.ijbiomac.2020.01.072. Epub 20200108.
- Lopes AH, Silva RL, Fonseca MD, Gomes FI, Maganin AG, Ribeiro LS, Marques LMM, Cunha FQ, Alves-Filho JC, Zamboni DS, et al. Molecular basis of carrageenan-induced cytokines production in macrophages. Cell Commun Signal. 2020;18:141. doi:10.1186/s12964-020-00621-x. Epub 20200907.
- Arslan R, Bor Z, Bektas N, Meriçli AH, Ozturk Y. Antithrombotic effects of ethanol extract of Crataegus orientalis in the carrageenan-induced mice tail thrombosis model. Thromb Res. 2011;127:210–3. doi:10.1016/j.thromres.2010.11.028. Epub 20101222.
- Barth CR, Funchal GA, Luft C, de Oliveira JR, Porto BN, Donadio MV. Carrageenan-induced inflammation promotes ROS generation and neutrophil extracellular trap formation in a mouse model of peritonitis. Eur J Immunol. 2016;46:964–70. doi:10.1002/eji.201545520. Epub 20160208.
- Manne BK, Getz TM, Hughes CE, Alshehri O, Dangelmaier C, Naik UP, Watson SP, Kunapuli SP. Fucoidan is a novel platelet agonist for the C-type lectin-like receptor 2 (CLEC-2). J Biol Chem. 2013;288:7717–26. doi:10.1074/jbc.M112.424473. Epub 20130122.
- Alshehri OM, Montague S, Watson S, Carter P, Sarker N, Manne BK, Miller JL, Herr AB, Pollitt AY, O’Callaghan CA, et al. Activation of glycoprotein VI (GPVI) and C-type lectin-like receptor-2 (CLEC-2) underlies platelet activation by diesel exhaust particles and other charged/hydrophobic ligands. Biochem J. 2015;468:459–73. doi:10.1042/bj20150192. Epub 20150407.
- Kardeby C, Fälker K, Haining EJ, Criel M, Lindkvist M, Barroso R, Påhlsson P, Ljungberg LU, Tengdelius M, Rainger GE, et al. Synthetic glycopolymers and natural fucoidans cause human platelet aggregation via PEAR1 and GPIbα. Blood Adv. 2019;3:275–87. doi:10.1182/bloodadvances.2018024950.
- Dul M, Paluch KJ, Kelly H, Healy AM, Sasse A, Tajber L. Self-assembled carrageenan/protamine polyelectrolyte nanoplexes-investigation of critical parameters governing their formation and characteristics. Carbohydr Polym. 2015;123:339–49. doi:10.1016/j.carbpol.2015.01.066. Epub 20150207.
- Martin EM, Zuidscherwoude M, Morán LA, Di Y, García A, Watson SP. The structure of CLEC-2: mechanisms of dimerization and higher-order clustering. Platelets. 2021;32:733–43. doi:10.1080/09537104.2021.1906407. Epub 20210405.
- Brown GD, Willment JA, Whitehead L. C-type lectins in immunity and homeostasis. Nat Rev Immunol. 2018;18:374–89. doi:10.1038/s41577-018-0004-8.
- Evaluations of the Joint FAO/WHO expert committee on food additives (JECFA). https://apps.who.int/food-additives-contaminants-jecfa-database/Home/Chemical/377.