
Abstract
Platelet-specific collagen receptor glycoprotein (GP)VI is stable on the surface of circulating platelets but undergoes ectodomain cleavage on activated platelets. Activation-dependent GPVI metalloproteolysis is primarily mediated by A Disintegrin And Metalloproteinase (ADAM) 10. Regulation of platelet ADAMs activity is not well-defined however Tissue Inhibitors of Metalloproteinases (TIMPs) may play a role. As levels of TIMPs on platelets and the control of ADAMs-mediated shedding by TIMPs has not been evaluated, we quantified the levels of TIMPs on the surface of resting and activated platelets from healthy donors by flow cytometry and multiplex ELISA. Variable levels of all TIMPs could be detected on platelets. Plasma contained significant quantities of TIMP1 and TIMP2, but only trace amounts of TIMP3 and TIMP4. Recombinant TIMP3 strongly ablated resting and activated platelet ADAM10 activity, when monitored using a quenched fluorogenic peptide substrate with ADAM10 specificity. Whilst ADAM10-specific inhibitor GI254023X or ethylenediamine tetraacetic acid (EDTA) could modulate ligand-initiated shedding of GPVI, only recombinant TIMP2 achieved a modest (~20%) inhibition. We conclude that some platelet TIMPs are able to modulate platelet ADAM10 activity but none strongly regulate ligand-dependent shedding of GPVI. Our findings provide new insights into the regulation of platelet receptor sheddase activity.
Plain Language Summary
What do we know?
Platelet receptor GPVI initiates platelet adhesion and aggregation and is proteolytically cleaved from the activated platelet surface
The metalloproteinases responsible belong to the ADAMs family of enzymes which are inhibited by TIMPs
What did we discover?
Plasma contains significant amounts of TIMP1 and TIMP2
Circulating platelets bear significant amounts of TIMPs 1, 2, and 3
Recombinant TIMP3 strongly inhibits resting and activated platelet ADAM10 activity
Exogenous addition of TIMP2 mildly blocked ligand-initiated shedding of GPVI
What is the impact?
TIMPs may modulate ADAM10 activity under resting conditions and stabilize GPVI levels in response to platelet activation
Anti-GPVI agents are being evaluated as anti-thrombotic agents, however, acute loss of GPVI in trauma or settings of thrombocytopenia is linked with clinical bleeding
Understanding how GPVI levels are regulated is important as agents that modulate GPVI function are emerging as important therapeutics for clinical applications in Thrombosis and Hemostasis fields
Introduction
Upon vascular injury or change in biorheological properties of blood, circulating platelets become activated through engagement of platelet surface molecules with extracellular matrix proteins.Citation1 The platelet-specific receptors glycoprotein (GP)Ib-IX-V and GPVI cooperate to mediate adhesion to von Willebrand factor (VWF) and to collagen, respectively.Citation2 Platelet activation triggers morphological and cytoskeletal changes and activation of signaling pathways resulting in fibrinogen binding to the integrin αIIbβ3. Metalloproteinase-mediated receptor shedding of the ectodomains of GPIbα and GPVI are also an important consequence of platelet activation.Citation3 GPVI is one of the several adhesive receptors that contribute to thrombus initiation,Citation4 and evidence that GPVI also binds fibrinogenCitation5 and fibrinCitation6,Citation7 suggest an important role for receptor proteolysis in thrombus stabilization.
On human platelets, surface levels of GPVI are regulated by A Disintegrin And Metalloproteinase (ADAM)10 to release a 55-kDa ectodomain fragment termed soluble GPVI (sGPVI).Citation8 ADAM17 cleaves the GPIbα subunit of GPIb-IX-V and also contributes to GPVI shedding in mice.Citation9 Shedding of GPVI can be induced rapidly via engagement by ligands,Citation10,Citation11 by engagement of platelet FcγRIIa by anti-platelet antibodies,Citation12 or by pathophysiological shear stress.Citation13 This irreversible release of the ligand-binding portion of GPVI provides a mechanism for downregulating platelet reactivity by altering receptor density. To date, there is no known biological function for the ~ 55-kDa sGPVI fragment. ADAM10 is ubiquitously expressed on mammalian cells and is constitutively active on the surface of platelets.Citation14 Activation-dependent metalloproteolysis is likely to represent a mechanism that regulates platelet responsesCitation15 as the relative surface densities of GPIbα and GPVI are important for control of platelet reactivity toward VWF, collagen, and other ligands in flowing blood.Citation16 However, the mechanisms by which platelet metalloproteinase activity is regulated, are not well understood.
Platelets express several ADAMs, including ADAM9, ADAM10 and ADAM17.Citation15,Citation17 ADAMs have major roles in the regulation and bioavailability of a host of growth factors, cytokines, and adhesion and cytokine receptors.Citation18,Citation19 In general, ADAMs metalloproteinases demonstrate minimal amino acid sequence selectivity for catalysisCitation20,Citation21 making these enzymes viable but challenging drug targets.Citation22 The enzymatic activity of ADAMs and other metalloproteinases is known to be modulated in part by a four-member Tissue Inhibitors of Metalloproteinase (TIMP) family.Citation23
The four TIMP family members are endogenous inhibitors of ADAMs and other metalloproteinases, and possess some redundancy in their abilities to inhibit specific metalloproteinases.Citation23,Citation24 Crystallographic studies revealed that the N-terminal region of human TIMPs interact with the catalytic domain of metalloproteinases in a 1:1 stoichiometry and inhibits proteolytic activity by disrupting the coordination of Zn2+ within the enzyme active site.Citation25,Citation26 Previous work in nucleated cells has revealed that ADAM10 is inhibited by TIMP1Citation27 and TIMP3,Citation28 while studies in vitro using recombinant human (rh) ADAM10 demonstrated that TIMP1 exhibited a markedly lower inhibition constant than TIMP3.Citation29 Several reports describe the expression of TIMPs in platelet lysates using techniques such as Western blotting and mass spectrometryCitation25,Citation30 and Villeneuve and colleaguesCitation31 observed sub-cellular localization of TIMPs in platelets by immunofluorescence. However, TIMP association with the platelet surface has not yet been explored.
Given the relative stability of GPVI on circulating platelets, despite catalytically-active ADAM10 being present on the platelet surface, this implies that there are other mechanisms that control ADAMs-mediated receptor shedding. Importantly, disruption of platelet–fibrin interactions mediated by GPVI (for example, by ectodomain shedding) has important implications for thrombus stability and embolization.Citation32 Anti-GPVI Fabs are emerging as viable anti-thrombotic agents with strong efficacy and reduced bleeding issues.Citation33,Citation34 Thus, it is important to elucidate molecular mechanisms that could potentially modulate human platelet GPVI levels. TIMPs could possibly contribute to the maintenance of platelet GPVI levels, by influencing platelet metalloproteinase activity. Here, we measured platelet TIMPs on resting and activated washed human platelets, in platelet-rich plasma (PRP) and levels of unbound TIMPs in plasma. We also assessed the influence of exogenous TIMPs on platelet ADAM10 activity and GPVI shedding in response to platelet activation, and platelet adhesion under flow. Platelets selectively bore TIMP3 on the surface, but the addition of exogenous TIMP3 did not protect GPVI from ligand-induced shedding. Our findings have important implications for targeting the regulation of GPVI in thrombosis and hemostasis.
Materials and methods
Specialist reagents
Rabbit polyclonal antibodies against human TIMP1, TIMP2, TIMP3, or TIMP4, as well as AlexaFluor488-labeled anti-mouse or anti-rabbit IgG raised in goats, were purchased from Abcam (Cambridge, UK). Purified recombinant human (rh) ADAM10, ADAM17, TIMP 1, TIMP2, TIMP3 or TIMP4 were purchased from R&D Systems (Minneapolis, MI, USA). Equine Type I collagen (Horm suspension) was from Takeda (Lincz, Austria). Convulxin was purchased from Enzo Life Sciences (Framingham, USA). Collagen-related peptide (CRP) from Auspep (Tullamarine, Australia) was chemically cross-linked (CRP-xL) in-house as described.Citation35
Human blood
Blood was collected from healthy individuals who were free of anticoagulant and antiplatelet medications, after provision of informed consent (Australian National University Human Research Ethics Committee protocol 2016/317). Venesection was performed using a 19-gauge winged infusion set, and either 3.2% (w/v) trisodium citrate (TSC) (9:1 ratio of blood to TSC) or 97 mM TSC containing 111 mM glucose and 78 mM citric acid (acid-citrate-dextrose; ACD) (6:1 ratio of blood to ACD) for PRP or washed platelet preparation, respectively. Whole blood was centrifuged at 110g for 20 min, at room temperature (RT) (Beckman Coulter Allegra X-12 R, Brea, California, USA) and the upper PRP layer was collected by aspiration. Washed platelets from PRP from ACD-anticoagulated blood were prepared by centrifugation at 1270g for 15 min, RT. Platelet-poor plasma (PPP) was removed and the platelet pellet was washed by gentle resuspension in 123 mM NaCl containing 13 mM TSC, pH 7.0, and 33.3 mM glucose (CGS buffer). Platelets were pelleted by centrifugation at 1270g for 15 min, RT and resuspended in CGS buffer twice then the platelet pellet was resuspended at 5 × 108 platelets/mL in 5.5 mM glucose, 137 mM NaCl, 12 mM NaHCO3, 1.8 mM CaCl2, 0.49 mM MgCl2.6 H2O, 2.7 mM KCl, 0.36 mM NaH2PO4.H2O, pH 7.4 (Tyrode’s buffer) to obtain washed platelets. Platelet numbers were enumerated using an automated analyzer (Abbott Diagnostics, Chicago, IL, USA).
Flow cytometry
Receptors on resting or activated washed platelets (5×108/mL) or PRP were assessed by flow cytometry. Platelet suspensions in 20 mM Tris-HCl containing 150 mM NaCl and 5 mM EDTA pH 7.4 (TS-EDTA) were incubated with 1 µg/mL AlexaFluor 488-conjugated anti-human GPVI monoclonal antibody, 1G5 (in-house), or 0.6 µg/mL PE-conjugated anti-CD41 monoclonal antibody (MEM-06, Abcam, Cambridge, UK), or 3 µg/mL anti-human TIMPs 1–4, or 0.6 µg/mL PE-conjugated mouse IgG1 isotype control (Abcam, Cambridge, UK), and incubated at RT for 30 min. For unconjugated primary antibodies, a further 30 min incubation with 1 µg/mL AlexaFluor 488-conjugated goat anti-mouse Ig or 2 µg/mL goat anti-rabbit Ig was performed at RT. Platelet suspensions were diluted by addition of 400 µL TS-EDTA and bound antibodies were analyzed by flow cytometry (LSR-Fortessa X-20, BD Biosciences, Franklin Lakes, New Jersey, USA). 10 000 platelet events were recorded and post-acquisition analysis was performed using FlowJo version 10.4 (FlowJo, LLC, Ashland, Oregon, USA).
SDS-Polyacrylamide electrophoresis and Western blotting
Purified recombinant TIMPs (0.2–0.4 µg) were mixed with 50 mM Tris-HCl, pH 6.8, 2% (w/v) sodium dodecyl sulfate (SDS), 10% (v/v) glycerol with or without 10 mM dithiothreitol and heated (100°C for 5 min) then centrifuged at 16 000g for 1 min before loading onto 10% acrylamide-bis gels. Proteins were resolved by electrophoresis, and then transferred to 0.2 µm polyvinylidene difluoride membranes (Amersham Hybond P, GE Healthcare, Chicago, IL, USA), via wet transfer (Trans-Blot Cell, Bio-Rad Laboratories, Hercules, California, USA) in 25 mM Tris-HCl pH 8.3 containing 190 mM glycine and 20% (v/v) methanol. Transelution was conducted at 100 V for 1 h, then membranes were blocked in 5% (w/v) skim milk in 20 mM Tris-HCl containing 137 mM NaCl and 0.1% (v/v) Tween-20 (TBS-T) for 1 h at RT or overnight at 4°C. TIMP bands were detected using 0.5–5 µg/mL primary antibody diluted in bovine serum albumin, for 1 h at RT or overnight at 4°C followed by 4X washes with TBS-T, then incubation with appropriate horseradish peroxidase-conjugated secondary antibody diluted 500–10,000-fold in TBS-T for 1 h. After washing, bound antibody was visualized using enhanced chemiluminescence substrate (GE Healthcare, Chicago, IL, USA) and quantified using Image Lab (v5.0 software; Bio-Rad ChemiDoc MP).
Quantitation of TIMPs in human plasma and serum
Levels of TIMPs 1–4 were quantified in double spun PPP and serum isolated from healthy donors using the magnetic bead-based immunoassay Human TIMP Magnetic Luminex Performance Assay 4-plex Kit according to manufacturer’s protocol. TIMP levels in standards of known TIMPs concentration together with duplicate samples were quantified using a MAGPIX analyzer with xPONENT 4.2 system (Luminex Corporation, Austin, Texas, USA). All values were corrected for nonspecific background signal.
ADAM10 activity assay
rhADAM10, rhADAM17, or human-washed platelet ADAM10 activity in the presence or absence of TIMPs 1–4 was measured using an established fluorescence resonance energy transfer (FRET) assay with GPVI-Cy3, a quenched fluorescent ADAM10-selective peptide sequence.Citation14 rhADAM10 or rhADAM17 (10–90 nM) were mixed with 1 µM GPVI-Cy3 substrate and made up to 100 µL with 0.1 M Tris-HCl pH 7.4 containing 150 mM NaCl, 5 µM ZnSO4 and 0.005% (v/v) Brij-35. Rates of ADAM10 activity were calculated by subtracting relative fluorescence unit (RFU) at time zero from RFU at 20 min.
Quantifying sGPVI
sGPVI levels in double spun PPP or supernatants collected from washed platelet experiments and treated with 50 mM (final concentration) EDTA were measured by an enzyme-linked immunosorbent assay (ELISA) as described.Citation36
Statistical analyses
Statistical analyses were performed using GraphPad Prism 9. Continuous variables were reported as mean ± SD.
Results
Verification of anti-TIMP antibody specificity
Levels of TIMP protein on platelets and mRNA in megakaryocytes have been previously assessed with variable findings regarding levels of specific isoforms.Citation31,Citation37,Citation38 As we wished to assess the potential for TIMPs to contribute to GPVI stability, we evaluated TIMP levels on resting and activated platelets by flow cytometry. First, we evaluated the specificity of commercial rabbit polyclonal anti-human TIMPs antibodies to be used in the study, for recognition of rhTIMPs by Western blot. Each membrane containing the four TIMP isoforms (molecular mass 22–25 kDa) were separately probed with saturating concentrations of each of the polyclonal anti-human TIMP antibodies. Figure S1 shows that the anti-hTIMP1 antibody did not detect any TIMPs, possibly due to loss of key epitope(s) within the denatured form of rhTIMP1. Anti-hTIMP2 antibody specifically recognized a sharp band at ~ 25 kDa corresponding to TIMP2 with no recognition of TIMP1, or TIMP3 and minimal cross-reactivity with TIMP4 (faint band at ~ 25 kDa). Likewise, anti-hTIMP3 antibody specifically detected TIMP3 with minor cross-reactivity with rhTIMP4 (faint band at ~ 22 kDa). A light band between 46 and 58 kDa was also recognized by anti-hTIMP3 antibody. Anti-hTIMP4 detected an ~ 22 kDa band and another band between 32 and 46 kDa of rhTIMP4. Some cross-reactivity with rhTIMP1 and rhTIMP3 may also be present (light band at ~ 22 kDa). On balance, apart from anti-hTIMP1, all the antibodies recognized the appropriate recombinant protein. Because platelet surface TIMPs were more relevant to subsequent studies evaluating ADAM10 activity and because all the TIMP antibodies have been demonstrated to perform well in flow cytometry assays,Citation39 it was decided to evaluate levels of TIMP proteins on resting and activated platelets by flow cytometry.
Assessment of TIMP levels on resting and activated platelets, and in plasma
Washed platelets from 4 to 6 healthy individuals were incubated separately with anti-hTIMP polyclonal antibodies and surface-associated antibodies were quantified by flow cytometry. Figure S2 shows example histogram traces from one healthy donor. A rightward shift in histogram peak for each TIMP isoform relative to control antibody was observed, suggesting all TIMP isoforms were detectable on washed platelets, with TIMP2 showing the greatest % positive events (48%; data not shown).
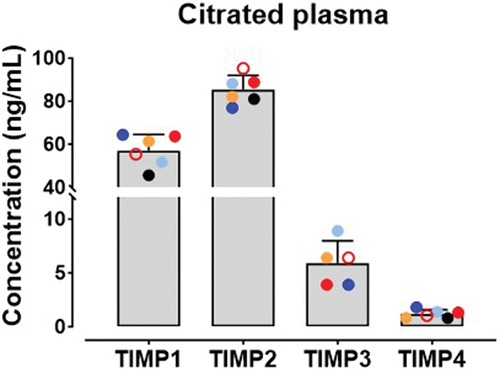
Platelet activation mediated by collagen–GPVI interactions triggers GPVI shedding. To assess TIMP levels on resting platelets and whether platelet activation resulted in changes to levels of TIMPs, washed platelets (5×108/mL) were treated with activating concentrations of collagen, CRP-xL, convulxin or Ca2+ ionophore sufficient to upregulate P-selectin levels on all donors (data not shown) and levels of TIMPs were assessed by flow cytometry. An incubation time of 2 h was identified as optimal for maximal detection of TIMP isoforms. Data in and Figure S3 show that levels of TIMPs on resting platelets did not change dramatically in platelets treated with collagen, CRP-xL or convulxin (p > .05), however, mild and donor-variable increases in TIMP levels (p = .0011 to p = .0187) were detected in Ca2+ ionophore-treated platelets. Similar modest TIMP upregulation was observed in PRP treated with Ca2+ ionophore but not collagen (data not shown). This suggests that like other cells, platelets store some TIMPs 1, 2 and 4 proteins which move to the surface under conditions of strong activation and may suggest that levels of TIMPs are dynamic on the platelet surface. All TIMPs could be detected in plasma from six healthy donors using a magnetic bead-based immunoassay (), with higher levels of TIMP1 and TIMP2 relative to TIMP3 and TIMP4. Among the isoforms, levels of TIMP2 was the highest at 85.39 ± 6.592 ng/mL followed by TIMP1 at 57.05 ± 7.497 ng/mL. Notably, less TIMP3 (5.90 ± 2.087 ng/mL) and TIMP4 (1.20 ± 0.382 ng/mL) were detected in plasma.
Figure 1. Levels of TIMPs on the surface of resting or activated platelets.
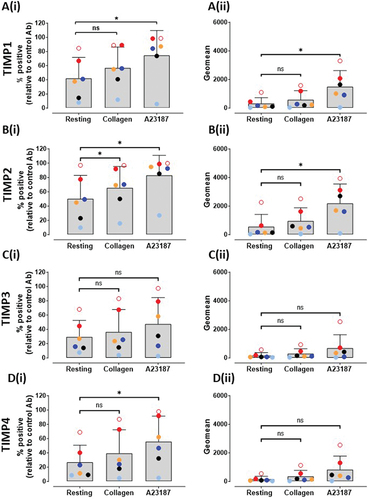
Figure 2. Levels of TIMPs in human plasma.
TIMP3 reduces resting and activated platelet ADAM10 activity
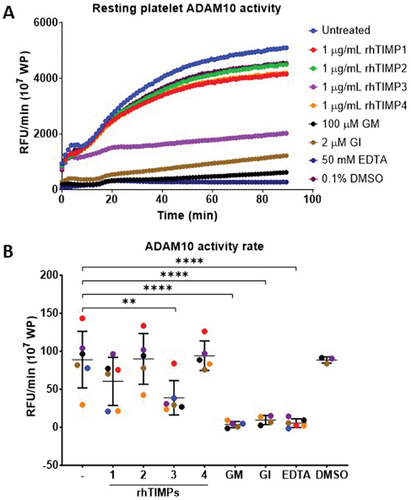
How platelet ADAM activity is regulated under resting and activated conditions has not been studied extensively. As TIMPs are endogenous inhibitors of ADAMs and human GPVI is predominantly cleaved by ADAM10, the relationship between TIMPs and platelet ADAM10 activity was explored using an established ADAM10 activity assay. As all four rhTIMP isoforms inhibited rhADAM10 activity to various extents (data not shown), we evaluated the effect of rhTIMPs on native platelet ADAM10 activity in washed platelets. Washed platelets (107/mL) were pre-treated with 1 µg/mL of rhTIMPs, 100 µM GM6001, 2 µM GI254023X, 50 mM EDTA or 0.1% (v/v) DMSO vehicle control for 30 min at RT before exposure to GPVI-Cy3 substrate. illustrates ADAM10 activity in 107 washed platelets from one healthy donor. Among the rhTIMP isoforms, only rhTIMP3 could inhibit platelet ADAM10 activity comparable with an ADAM10-specific inhibitor (2 µM GI254023X), a broad-spectrum metalloproteinase inhibitor (100 µM GM6001) or 50 mM EDTA. The rates of ADAM10 activity on resting platelets were variable among six healthy donors at 88.78 ± 37.23 RFU/min. Consistent with the effect on rhADAM10, rhTIMP3 significantly reduced resting platelet ADAM10 activity rate by 2.3-fold at 38.76 ± 22.66 RFU/min (p = .0054). Platelet ADAM10 activity was essentially abolished (p < .0001) by metalloproteinase inhibitors whereby a 24.6-fold, 9.4-fold and 16.6-fold reduction in rate of activity was observed with 100 µM GM6001, 2 µM GI254023X and 50 mM EDTA, respectively (). The inhibition of resting platelet ADAM activity by rhTIMP3 represented 63% of that obtained using the ADAM10-specific inhibitor GI254023X, implying that TIMP3 could potentially modulate resting platelet ADAM10 activity. In contrast, the mean rate of proteolysis of platelet ADAM10 remained stable in the presence of rhTIMP1, rhTIMP2, or rhTIMP4.
Figure 3. TIMP3 selectively inhibits resting platelet ADAM10 activity.
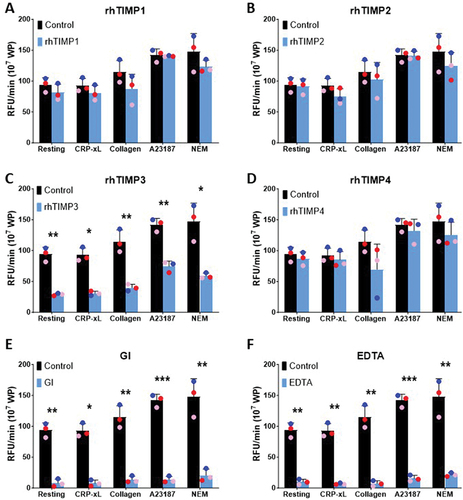
Under conditions of platelet activation, TIMP-mediated control of platelet ADAM10 activity may be altered. Thus, washed platelets (107) from healthy donors were pre-treated with 1 µg/mL of rhTIMPs, 2 µM GI254023X, or 50 mM EDTA for 30 min at RT before exposure to GPVI-Cy3 substrate in the presence of 10 µg/mL CRP-xL, 10 µg/mL collagen, 10 µM calcium ionophore (A23187) or 5 mM NEM, a potent activator of ADAM10. Among the four rhTIMP isoforms tested, only rhTIMP3 achieved a statistically significant reduction in ADAM10 activity rate in activated platelets (). On exposure to GPVI agonists, Ca2+ ionophore, or a potent activator of ADAM10, pre-treatment of platelets with rhTIMP3 significantly lowered the rate of ADAM10 activity by 1.9- to 3.2-fold compared to untreated platelets (p < .05, ), consistent with the effect observed on resting platelets (). However, the extent of ADAM10 inhibition by rhTIMP3 was inferior to the ADAM10-specific inhibitor GI254023X () or EDTA (). Platelets treated with GI254023X () showed a 7.2- to 12.0-fold lower rate of ADAM10 activity on activated platelets while the inclusion of EDTA () reduced ADAM10 rate of activity by 6.9- to 15.9-fold. Taken together, the data suggest that exogenous addition of rhTIMP3 significantly reduced platelet ADAM10 activity.
Figure 4. TIMP3 selectively inhibits activated platelet ADAM10 activity.
Exogenous recombinant TIMPs do not strongly reduce ligand-GPVI shedding
Inhibition of platelet ADAM10 activity under resting and activated conditions appeared to be a feature of rhTIMP3, and so we evaluated the extent to which TIMPs could influence ADAM10-induced release of the platelet endogenous substrate, sGPVI. Washed platelets from three individual healthy donors were pre-treated with either 1 µg/mL of rhTIMP1–4, 2 µM GI254023X, or 50 mM EDTA for 30 min at RT. Platelets were subsequently stimulated with 10 ng/mL convulxin, 30 µg/mL CRP-xL, 10 µg/mL collagen or 10 µM Ca2+ ionophore for 2 h at RT, which have been shown to be conditions that cause activation of GPVI metalloproteolysis. Levels of sGPVI in the platelet supernatant was quantified by ELISA. Activation of washed platelets increased sGPVI levels from 12.09 ± 0.88 ng/mL in resting platelets to 46.93 ± 4.84 ng/mL with convulxin, 35.09 ± 8.26 ng/mL with CRP-xL, 23.06 ± 5.57 ng/mL with collagen and 137.88 ± 4.01 ng/mL with Ca2+ ionophore treatments (). Surprisingly, pre-treatment with rhTIMP3 did not alter the release of sGPVI from activated platelets. An ~ 20% reduction in convulxin-triggered sGPVI release was achieved by rhTIMP2 (p = .0059, ) while rhTIMP4 enhanced the release of sGPVI triggered by Ca2+ ionophore by 1.1-fold (p = .0322, ). confirms that ADAM10 was responsible for shedding of GPVI induced by GPVI agonists or Ca2+ ionophore as GI254023X inhibited sGPVI release by > 90% or 1.5- to 4.7-fold (p < .05), depending on the agonist used. EDTA markedly reduced the levels of sGPVI liberated by greater than 2-fold in all conditions of platelet activation (). Taken together, these data suggest that whilst exogenously added TIMP3 could ablate platelet ADAM10 activity, under conditions of ligand-induced shedding, only TIMP2 was able to mildly diminish ADAM10-mediated cleavage of GPVI. Furthermore, no significant relationship between plasma TIMP or platelet TIMP levels (PRP) with sGPVI could be demonstrated in samples from 6 healthy donors (). TIMP levels as assessed by antibody binding to washed platelets correlated with sGPVI levels, possibly implying that TIMPs may behave as markers of platelet activation. However, this idea warrants further investigation in a larger cohort to assess changes in the levels of TIMPs upon platelet activation.
Figure 5. Exogenously added rhTIMP isoforms minimally inhibit release of sGPVI from activated platelets.
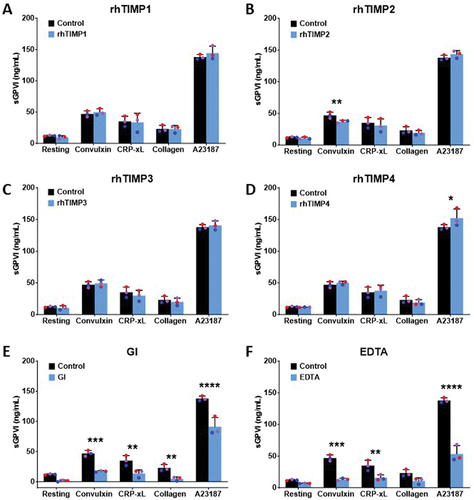
Table I. Relationship between platelet and plasma TIMPs and sGPVI levels in healthy individuals.
Discussion
In this study, we evaluated platelet surface-associated TIMPs and investigated whether TIMPs could regulate platelet ADAM10 activity. The antibodies used in our study were assessed for isoform specificity by Western blot. All antibodies except for the anti-TIMP1 antibody performed well and showed appropriate isoform selectivity. The TIMP1 antibody did not identify any recombinant TIMP isoform by Western blot, most likely due to an absence of TIMP1 epitope in the recombinant TIMP1 protein. This antibody has been reported to recognize TIMP1 by Western blot of lysed bronchial epithelial cells.Citation40 All four TIMP isoforms were detectable by flow cytometry on washed platelets and PRP under resting conditions consistent with the detection of TIMP1, TIMP2, and TIMP3 in platelet lysates by mass spectrometry.Citation41 We found that TIMP4 was detected at low levels by flow cytometry. TIMP4 was not detected in the platelet proteome,Citation41 but was strongly detected via Western blot analysis of platelet lysates.Citation37 The lack of concordance in TIMP4 detection in previous studies may be related to differences in the detection technique or antibody used. In line with the relative abundance of TIMP transcripts in platelets (TIMP1 > TIMP2 > TIMP3 > TIMP4) reported by Cecchetti and colleagues,Citation38 higher levels of surface TIMP1 and TIMP2 were observed compared to surface TIMP3 and TIMP4 on washed platelets and PRP. Intriguingly, TIMP4 mRNA was absent in the platelet transcriptome despite low levels of TIMP4 transcript detected in megakaryocytes.Citation38 Detection of TIMP4 on the platelet surface, within sub-cellular platelet compartmentsCitation31 and in platelet lysateCitation37 may suggest the ability of platelets to take up TIMPs from plasma and/or transfer of TIMPs from megakaryocyte progenitors.
All four TIMP isoforms were more readily detectable on washed platelets compared to PRP, and a positive correlation between sGPVI and TIMP levels on washed platelets was identified. In line with the increase of TIMPs observed on activated platelets, TIMPs may behave as markers of platelet activation. The plasma concentrations of each TIMP isoform in samples from healthy individuals in our study were comparable to previous reports.Citation42–45 However, the variation in platelet-associated TIMPs between donors were smaller in PRP than in washed platelet samples, possibly reflecting binding properties of each TIMP in plasma relative to the membrane-bound TIMP, serving to reduce the variability in surface TIMP levels observed in PRP. Future studies should incorporate relevant donor features relating to diet, medicines prescribed or thrombosis risk factors, all of which might explain variable levels of TIMPs amongst healthy donors.
Levels of platelet TIMPs were increased in response to Ca2+ ionophore (A23187) treatment, that also caused platelet degranulation. Villeneuve and colleaguesCitation31 observed an upregulation of all four TIMP isoforms in the supernatant of thrombin-activated platelets, while Fong and colleaguesCitation30 reported the release of TIMP1, TIMP2, and TIMP3 from phorbol myristate acetate-activated platelets. When considered together with our findings, treatments that result in platelet degranulation may mobilize an intracellular store of TIMPs to the membrane surface. Activated platelets increase exposure of P-selectin, phosphatidylserine as well as CD63, a dense granule GP reported to bind TIMP1.Citation46 As there was no correlation evident between plasma and surface TIMPs (data not shown), a level of control of TIMP localization may exist, preventing the respective TIMP levels from reaching an equilibrium.
We used saturating amounts (1 µg/mL) of each rhTIMP to comprehensively assess capacity to inhibit platelet ADAM10 activity. Exogenously added rhTIMP3 reduced both resting and activated platelet ADAM10 activity, while there was a trend to reduced resting platelet ADAM10 activity with exogenous rhTIMP1. This observation is supported by the inhibitory effect of rhTIMP1 and rhTIMP3 on rhADAM10 activity reported elsewhere.Citation29 The unique inhibitory capacity of rhTIMP3 on activated platelet ADAM10 activity may reflect molecular differences in conformation of the TIMP isoforms. Crystallography studies by Wisniewska and colleaguesCitation47 revealed that the N-terminal domain of TIMP3 is more tightly packed into the catalytic site of ADAM17, a second ADAM family member with strong homology to ADAM10. This structural feature of TIMP3 may allow favorable interactions and confer greater binding efficiency, explaining data presented here whereby TIMP3 appeared to be the most effective TIMP isoform in inhibiting platelet ADAM10 activity.
Although rhTIMP3 exhibited an inhibitory effect on ADAM10 activity, the reduced ADAM10 activity did not affect activation-dependent sGPVI release. In healthy donor-washed platelets, rhTIMP3 as well as TIMP1 and TIMP4 did not protect GPVI from shedding upon stimulation with convulxin, collagen, CRP-xL or calcium ionophore despite a > 80% reduction in platelet ADAM10 activity by rhTIMP3 in the presence of these agonists. However, release of GPVI from activated platelets was blocked by inclusion of a small molecule ADAM10 inhibitor, GI254023X (391.5 Da) or EDTA (292.2 Da). By comparison, the relatively larger rhTIMP3 (24 kDa) may hinder its access to the catalytic region of ADAM10 under conditions of rapid activation. Further, it is possible that additional membrane factors also control access of ADAM10 to the GPVI cleavage site. Several studies have identified roles for tetraspanins in regulating ADAM10 metalloproteolysis and GPVI release.Citation15,Citation48,Citation49 The discordance between ADAM10 activity and the release of sGPVI agrees with previous workCitation14 showing increased release of sGPVI can occur without an increase in the catalytic activity of platelet ADAM10 upon activation with GPVI ligands, suggesting ligand-induced structural changes to GPVI and ADAM10 enhance access of the ADAM10 catalytic site for GPVI. However, an increase in ADAM10 activity driven by exposure to non-ligand platelet agonists such as fluid shear stress, calmodulin inhibitors or NEM, resulted in a corresponding increase in sGPVI.Citation14 Hence, it would be interesting to investigate the effect of rhTIMP3 on ADAM10 activity and sGPVI release in response to these ligand-independent agonists.
The data presented here suggest that dynamic platelet-associated levels of TIMPs may modulate ADAM10 activity, for example under resting conditions and maintain platelet patency by stabilizing receptor levels in response to platelet activation. Considering the role of GPVI in thrombus initiation, regulation of platelet ADAM10 by TIMPs may have implications for thrombus formation. Understanding the mechanistic basis of ADAM10 regulationCitation50 may open therapeutic avenues in managing thrombotic and bleeding conditions. Future work should assess how TIMPs impact ADAM10 activity and GPVI surface levels in models of thrombus formation.
Author contributions
CSML, AK, SJM and SMH performed experiments; CSML analyzed data and wrote the original manuscript draft. RKA and EEG designed the study and wrote the manuscript. All authors interpreted results and reviewed the manuscript.
Acknowledgments
The authors thank their colleagues for helpful discussions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hearn JI, Gardiner EE. Research and clinical approaches to assess platelet function in flowing blood. Arterioscler Thromb Vasc Biol. 2023;43(10):1775–10. doi:10.1161/ATVBAHA.123.317048.
- Quach ME, Li R. Structure-function of platelet glycoprotein Ib-IX. J Thromb Haemost. 2020;18(12):3131–41. doi:10.1111/jth.15035.
- Gardiner EE. Proteolytic processing of platelet receptors. Res Pract Thromb Haemost. 2018;2:240–50. doi:10.1002/rth2.12096.
- Perrella G, Nagy M, Watson SP, Heemskerk JWM. Platelet GPVI (glycoprotein VI) and thrombotic complications in the venous system. Arterioscler Thromb Vasc Biol. 2021;41(11):2681–92. doi:10.1161/ATVBAHA.121.316108.
- Mangin PH, Onselaer MB, Receveur N, Le Lay N, Hardy AT, Wilson C, Sanchez X, Loyau S, Dupuis A, Babar AK, et al. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica. 2018;103(5):898–907. doi:10.3324/haematol.2017.182972.
- Mammadova-Bach E, Ollivier V, Loyau S, Schaff M, Dumont B, Favier R, Freyburger G, Latger-Cannard V, Nieswandt B, Gachet C, et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood. 2015;126(5):683–91. doi:10.1182/blood-2015-02-629717.
- Alshehri OM, Hughes CE, Montague S, Watson SK, Frampton J, Bender M, Watson SP. Fibrin activates GPVI in human and mouse platelets. Blood. 2015;126(13):1601–8. doi:10.1182/blood-2015-04-641654.
- Gardiner EE, Karunakaran D, Shen Y, Arthur JF, Andrews RK, Berndt MC. Controlled shedding of platelet glycoprotein (GP)VI and GPIb–IX–V by ADAM family metalloproteinases. J Thromb Haemost. 2007;5(7):1530–7. doi:10.1111/j.1538-7836.2007.02590.x.
- Bender M, Hofmann S, Stegner D, Chalaris A, Bösl M, Braun A, Scheller J, Rose-John S, Nieswandt B. Differentially regulated GPVI ectodomain shedding by multiple platelet–expressed proteinases. Blood. 2010;116(17):3347. doi:10.1182/blood-2010-06-289108.
- Gardiner EE, Arthur JF, Kahn ML, Berndt MC, Andrews RK. Regulation of platelet membrane levels of glycoprotein VI by a platelet-derived metalloproteinase. Blood. 2004;104(12):3611–7. doi:10.1182/blood-2004-04-1549.
- Montague SJ, Hicks SM, Lee CS, Coupland LA, Parish CR, Lee WM, Andrews RK, Gardiner EE. Fibrin exposure triggers αIIbβ3-independent platelet aggregate formation, ADAM10 activity and glycoprotein VI shedding in a charge-dependent manner. J Thromb Haemost. 2020;18(6):1447–58. doi:10.1111/jth.14797.
- Gardiner EE, Al-Tamimi M, Mu FT, Karunakaran D, Thom JY, Moroi M, Andrews RK, Berndt MC, Baker RI. Compromised ITAM-based platelet receptor function in a patient with immune thrombocytopenic purpura. J Thromb Haemostasis. 2008;6(7):1175–82. doi:10.1111/j.1538-7836.2008.03016.x.
- Al-Tamimi M, Tan C, Qiao J, Pennings GJ, Javadzadegan A, Yong ASC, Arthur JF, Davis AK, Jing J, Mu FT, et al. Pathological shear triggers shedding of vascular receptors: a novel mechanism for downregulation of platelet glycoprotein (GP)VI in stenosed coronary vessels. Blood. 2012;119:4311–20. doi:10.1182/blood-2011-10-386607.
- Facey A, Pinar I, Arthur JF, Qiao J, Jing J, Mado B, Carberry J, Andrews RK, Gardiner EE. A-disintegrin-and-metalloproteinase (ADAM)10 activity on resting and activated platelets. Biochemistry. 2016;55(8):1187–94. doi:10.1021/acs.biochem.5b01102.
- Matthews AL, Noy PJ, Reyat JS, Tomlinson MG. Regulation of a disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: the emerging role of tetraspanins and rhomboids. Platelets. 2017;28(4):333–41. doi:10.1080/09537104.2016.1184751.
- Kruss S, Erpenbeck L, Amschler K, Mundinger TA, Boehm H, Helms H-J, Friede T, Andrews RK, Schön MP, Spatz JP. Adhesion maturation of neutrophils on nanoscopically presented platelet glycoprotein Ibα. Acs Nano. 2013;7(11):9984–96. doi:10.1021/nn403923h.
- Santos-Martinez MJ, Medina C, Jurasz P, Radomski MW. Role of metalloproteinases in platelet function. Thromb Res. 2008;121(4):535–42. doi:10.1016/j.thromres.2007.06.002.
- Lichtenthaler SF, Lemberg MK, Fluhrer R. Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J. 2018;37:e99456. doi:10.15252/embj.201899456.
- Giebeler N, Zigrino P. A disintegrin and metalloprotease (ADAM): historical overview of their functions. Toxins. 2016;8(4):122. doi:10.3390/toxins8040122.
- Maurer S, Kopp HG, Salih HR, Kropp KN. Modulation of immune responses by platelet-derived ADAM10. Front Immunol. 2020;11:44. doi:10.3389/fimmu.2020.00044.
- Lorenzen I, Lokau J, Korpys Y, Oldefest M, Flynn CM, Künzel U, Garbers C, Freeman M, Grötzinger J, Düsterhöft S. Control of ADAM17 activity by regulation of its cellular localisation. Sci Rep. 2016;6(1):35067. doi:10.1038/srep35067.
- Camodeca C, Cuffaro D, Nuti E, Rossello A. ADAM metalloproteinases as potential drug targets. Curr Med Chem. 2019;26(15):2661–89. doi:10.2174/0929867325666180326164104.
- Jackson HW, Defamie V, Waterhouse P, Khokha R. Timps: versatile extracellular regulators in cancer. Nat Rev Cancer. 2017;17(1):38–53. doi:10.1038/nrc.2016.115.
- Gresele P, Falcinelli E, Sebastiano M, Momi S. Matrix metalloproteinases and platelet function. Prog Mol Biol Transl Sci. 2017;147:133–65.
- Murphy G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011;12(11):233–40. doi:10.1186/gb-2011-12-11-233.
- Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol. 2013;13(9):649–65. doi:10.1038/nri3499.
- Schelter F, Grandl M, Seubert B, Schaten S, Hauser S, Gerg M, Boccaccio C, Comoglio P, Kruger A. Tumor cell-derived TIMP-1 is necessary for maintaining metastasis-promoting met-signaling via inhibition of ADAM-10. Clin Exp Metastasis. 2011;28(8):793–802. doi:10.1007/s10585-011-9410-z.
- Scilabra SD, Pigoni M, Pravata V, Schatzl T, Muller SA, Troeberg L, Lichtenthaler SF. Increased TIMP-3 expression alters the cellular secretome through dual inhibition of the metalloprotease ADAM10 and ligand-binding of the LRP-1 receptor. Sci Rep. 2018;8(1):14697. doi:10.1038/s41598-018-32910-4.
- Amour A, Knight CG, Webster A, Slocombe PM, Stephens PE, Knauper V, Docherty AJ, Murphy G. The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 2000;473(3):275–9. doi:10.1016/S0014-5793(00)01528-3.
- Fong KP, Barry C, Tran AN, Traxler EA, Wannemacher KM, Tang H-Y, Speicher KD, Blair IA, Speicher DW, Grosser T, et al. Deciphering the human platelet sheddome. Blood. 2011;117:15–26. doi:10.1182/blood-2010-05-283838.
- Villeneuve J, Block A, Le Bousse-Kerdiles MC, Lepreux S, Nurden P, Ripoche J, Nurden AT. Tissue inhibitors of matrix metalloproteinases in platelets and megakaryocytes: a novel organization for these secreted proteins. Exp Hematol. 2009;37(7):849–56. doi:10.1016/j.exphem.2009.03.009.
- Ahmed MU, Kaneva V, Loyau S, Nechipurenko D, Receveur N, Le Bris M, Janus-Bell E, Didelot M, Rauch A, Susen S, et al. Pharmacological blockade of glycoprotein VI promotes thrombus disaggregation in the absence of thrombin. Arterioscler Thromb Vasc Biol. 2020;40(9):2127–42. doi:10.1161/ATVBAHA.120.314301.
- Billiald P, Slater A, Welin M, Clark JC, Loyau S, Pugnière M, Jiacomini IG, Rose N, Lebozec K, Toledano E, et al. Targeting platelet GPVI with glenzocimab: a novel mechanism for inhibition. Blood Adv. 2023;7:1258–68. doi:10.1182/bloodadvances.2022007863.
- Wichaiyo S, Parichatikanond W, Rattanavipanon W. Glenzocimab: A GPVI (Glycoprotein VI)-targeted potential antiplatelet agent for the treatment of acute ischemic stroke. Stroke. 2022;53(11):3506–13. doi:10.1161/STROKEAHA.122.039790.
- Morton LF, Hargreaves PG, Farndale RW, Young RD, Barnes MJ. Integrin α2β1-independent activation of platelets by simple collagen-like peptides: collagen tertiary (triple-helical) and quaternary (polymeric) structures are sufficient alone for α2β1-independent platelet reactivity. Biochem J. 1995;306:337–44. doi:10.1042/bj3060337.
- Al-Tamimi M, Mu F-T, Moroi M, Gardiner EE, Berndt MC, Andrews RK. Measuring soluble platelet glycoprotein VI in human plasma by ELISA. Platelets. 2009;20(3):143–9. doi:10.1080/09537100802710286.
- Radomski A, Jurasz P, Sanders EJ, Overall CM, Bigg HF, Edwards DR, Radomski MW. Identification, regulation and role of tissue inhibitor of metalloproteinases-4 (TIMP-4) in human platelets. Br J Pharmacol. 2002;137(8):1330–8. doi:10.1038/sj.bjp.0704936.
- Cecchetti L, Tolley ND, Michetti N, Bury L, Weyrich AS, Gresele P. Megakaryocytes differentially sort mRNAs for matrix metalloproteinases and their inhibitors into platelets: a mechanism for regulating synthetic events. Blood. 2011;118(7):1903–11. doi:10.1182/blood-2010-12-324517.
- Clark RT, Nance JP, Noor S, Wilson EH. T-cell production of matrix metalloproteinases and inhibition of parasite clearance by TIMP-1 during chronic toxoplasma infection in the brain. ASN Neuro. 2011;3:e00049. doi:10.1042/AN20100027.
- Li P, Wang J, Guo F, Zheng B, Zhang X. A novel inhibitory role of microRNA-224 in particulate matter 2.5-induced asthmatic mice by inhibiting TLR2. J Cell Mol Med. 2020;24(5):3040–52. doi:10.1111/jcmm.14940.
- Burkhart JM, Vaudel M, Gambaryan S, Radau S, Walter U, Martens L, Geiger J, Sickmann A, Zahedi RP. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120(15):e73–82. doi:10.1182/blood-2012-04-416594.
- Palei AC, Sandrim VC, Cavalli RC, Tanus-Santos JE. Comparative assessment of matrix metalloproteinase (MMP)-2 and MMP-9, and their inhibitors, tissue inhibitors of metalloproteinase (TIMP)-1 and TIMP-2 in preeclampsia and gestational hypertension. Clin Biochem. 2008;41(10–11):875–80. doi:10.1016/j.clinbiochem.2008.04.015.
- Su C-W, Su B-F, Chiang W-L, Yang S-F, Chen M-K, Lin C-W. Plasma levels of the tissue inhibitor matrix metalloproteinase-3 as a potential biomarker in oral cancer progression. Int J Med Sci. 2017;14(1):37–44. doi:10.7150/ijms.17024.
- Hendrickson CM, Gibb SL, Miyazawa BY, Keating SM, Ross E, Conroy AS, Calfee CS, Pati S, Cohen MJ. Elevated plasma levels of TIMP-3 are associated with a higher risk of acute respiratory distress syndrome and death following severe isolated traumatic brain injury. Trauma Surg Acute Care Open. 2018;3:e000171. doi:10.1136/tsaco-2018-000171.
- Cavusoglu E, Kassotis JT, Marmur JD, Banerji MA, Yanamadala S, Chopra V, Anwar A, Eng C. Usefulness of plasma tissue inhibitor of matrix metalloproteinase-4 to predict death and myocardial infarction in patients with diabetes mellitus referred for coronary angiography. Am J Cardiol. 2017;120(1):1–7. doi:10.1016/j.amjcard.2017.03.267.
- Jung K-K, Liu X-W, Chirco R, Fridman R, Kim H-RC. Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. 2006;25(17):3934–42. doi:10.1038/sj.emboj.7601281.
- Wisniewska M, Goettig P, Maskos K, Belouski E, Winters D, Hecht R, Black R, Bode W. Structural determinants of the ADAM inhibition by TIMP-3: crystal structure of the TACE-N-TIMP-3 complex. J Mol Biol. 2008;381(5):1307–19. doi:10.1016/j.jmb.2008.06.088.
- Koo CZ, Matthews AL, Harrison N, Szyroka J, Nieswandt B, Gardiner EE, Poulter NS, Tomlinson MG. The platelet collagen receptor GPVI is cleaved by Tspan15/ADAM10 and Tspan33/ADAM10 molecular scissors. Int J Mol Sci. 2022;23(5):2440. doi:10.3390/ijms23052440.
- Eschenbrenner E, Jouannet S, Clay D, Chaker J, Boucheix C, Brou C, Tomlinson MG, Charrin S, Rubinstein E. TspanC8 tetraspanins differentially regulate ADAM10 endocytosis and half-life. Life Sci Alliance. 2020;3:e201900444. doi:10.26508/lsa.201900444.
- Seegar TCM, Killingsworth LB, Saha N, Meyer PA, Patra D, Zimmerman B, Janes PW, Rubinstein E, Nikolov DB, Skiniotis G, et al. Structural basis for regulated proteolysis by the alpha-secretase ADAM10. Cell. 2017;171:1638–48 e7. doi:10.1016/j.cell.2017.11.014.