
Abstract
Receptor-induced tyrosine phosphorylation of spleen tyrosine kinase (Syk) has been studied extensively in hematopoietic cells. Metabolic mapping and high-resolution mass spectrometry, however, indicate that one of the most frequently detected phosphorylation sites encompassed S297 (S291 in mice) located within the linker B region of Syk. It has been reported that Protein kinase C (PKC) phosphorylates Syk S297, thus influencing Syk activity. However, conflicting studies suggest that this phosphorylation enhances as well as reduces Syk activity. To clarify the function of this site, we generated Syk S291A knock-in mice. We used platelets as a model system as they possess Glycoprotein VI (GPVI), a receptor containing an immunoreceptor tyrosine-based activation motif (ITAM) which transduces signals through Syk. Our analysis of the homozygous mice indicated that the knock-in platelets express only one isoform of Syk, while the wild-type expresses two isoforms at 69 and 66 kDa. When the GPVI receptor was activated with collagen-related peptide (CRP), we observed an increase in functional responses and phosphorylations in Syk S291A platelets. This potentiation did not occur with AYPGKF or 2-MeSADP, although they also activate PKC isoforms. Although there was potentiation of platelet functional responses, there was no difference in tail bleeding times. However, the time to occlusion in the FeCl3 injury model was enhanced. These data indicate that the effects of Syk S291 phosphorylation represent a significant outcome on platelet activation and signaling in vitro but also reveals its multifaceted nature demonstrated by the differential effects on physiological responses in vivo.
Plain Language Summary
What is the context
Spleen tyrosine kinase (Syk) is present a number of cells and important in controlling the functions of various cells and organs.
Syk is known to exist in two isoforms Syk L (long form or Syk A) and Syk S (short form or Syk B).
It is known that phosphorylation events regulate Syk activation and activity.
In several inflammatory disease conditions, Syk mutants are known to play a role.
Phosphorylation of the Syk residue Serine 291 is known to occur, but its function in the regulation of Syk activation or activity is not known.
What is new
In this study, we generated a mutant mouse Syk S291A, which cannot be phosphorylated on serine residue. We evaluated the function of platelets isolated from these mice and compared them to platelets isolated from wild type littermates.
We observed that the mutation in Syk L unexpectedly caused Syk S to disappear from a number of tissues.
Platelet functions are enhanced in mutant mouse platelets compared to those from wild-type mice.
What is the impact
These studies enhance our understanding of the impact of Serine 291 phosphorylation on the function of Syk in platelets.
Introduction
Spleen tyrosine kinase (Syk) plays a critical role in a system that couples immunoreceptor tyrosine-based activation motifs (ITAMs) to intracellular signal transduction pathways in hematopoietic cells such as B cells, mast cells and platelets.Citation1 Syk activation initiates multiple signaling cascades which results in an array of inflammatory or immunological outcomes.Citation2 Upon ligand-induced activation, Syk is recruited to a pair of phosphorylated tyrosines in the ITAM, via its tandem Src homology 2 (SH2) domains in the N-terminal half of this kinase.Citation2–4 For example, in mouse platelets, ITAM-mediated Syk activation occurs through the Glycoprotein VI (GPVI)/FcR γ-chain receptor. Upon GPVI/FcR γ chain engagement, Src-family tyrosine protein kinases (SFKs) phosphorylate the ITAM resulting in the recruitment of Syk to the receptor complex. This leads to the activation of Syk and phosphorylation of its tyrosines that control Syk activation and interactions with downstream effectors, including LAT and PLCγ2. In vivo, Syk has been reported to be essential in maintaining vascular integrity and wound healing. Syk -/- mice displayed petechiae, endured severe hemorrhaging and demonstrated perinatal lethality.Citation5,Citation6
Besides the full-length isoform of Syk, a shorter splice variant form that lacks a 23-amino acid-long stretch in the interdomain B region as a result of exon 7 of the syk gene being spliced out is also expressed in humans and rodents.Citation5–9 Several nomenclature systems exist to denote these isoforms; they will be termed here SykL (long) and SykS (short) for consistency’s sake. In human cells, these two isoforms have been shown to have differential expression, sub-cellular localization, and functional effects in cancer cells,Citation1,Citation2,Citation8,Citation10,Citation11 suggesting the existence of alternative splicing-dependent mechanisms operating in concert with other mechanisms controlling Syk activity. Several studies have identified differential functions of Syk isoforms in cancer progression and metastasis,Citation12–14 including such fundamental cellular phenomena like mitosis, cell cycle, motility, apoptosis and survival.Citation15–17
Receptor-induced Syk tyrosine phosphorylation has been extensively studied.Citation1–Citation2–20 Through a combination of phosphopeptide mapping and mass-spectrometric analysis, Paris et al.Citation21 found that Syk S291 (S297 in human) was a major site of phosphorylation after murine B-cell receptor (BCR) engagement and that protein kinase C phosphorylated this site. Substitution of S291 with Ala decreased Syk-mediated BCR signaling but did not affect its activity.Citation21 This substitution, however, decreased Syk-mediated BCR signaling to activate NFAT and ELK-1. Later, Bohnenberger et al.Citation22 using high-resolution mass spectrometry identified Syk S297 as one of the most prominent phosphorylation sites of the B-lymphoid interactome of human Syk. This group reported that Syk can be associated with more than 25 distinct ligands including the 14-3-3γ adaptor protein which binds directly to phosphoserine 297. This complex reduced plasma membrane recruitment of Syk, thus limiting downstream signaling. As the SykS lacks the S291 containing region, which is encoded by exon 7, these two isoforms likely bind to different signaling molecules and may serve as nodal points for alternate signaling pathways.
In B lymphocytes, Akt was shown to be the kinase phosphorylating S297 and regulating its activity.Citation23 We have shown that a pan-PKC inhibitor affects Syk tyrosine phosphorylation and downstream signaling events in human platelets but not in murine platelets.Citation24 More recently, Makhoul et al.Citation25 demonstrated that ITAM-activated Syk caused PKC-dependent phosphorylation of Syk S297 in human platelets, which is consistent with the observations of Paris et al.Citation21 that PKC is the kinase that phosphorylates Syk S297. The authors concluded that Syk S297 phosphorylation represented a mechanism for feedback inhibition in human platelets.
To further our knowledge of the role Syk S297 phosphorylation plays in cell activation and downstream signaling, we generated Syk (S291A) knock-in mice. Our analysis of the homozygous mice indicated that the knock-in platelets express only one isoform of Syk, while the wild-type expresses two isoforms at 69 and 66 kDa. We also demonstrate that this mutation caused an increase in GPVI-induced aggregation and ATP secretion as well as the potentiation of phosphorylation of numerous tyrosine sites on Syk. Downstream signaling, measured by LAT and PLCγ2 phosphorylation, was also enhanced in the Syk (S291A) mouse platelets compared to their wild-type littermates. There was no effect of this mutation on bleeding times as measured by tail clipping, although there was a statistically significant reduction in the time to thrombus formation and vessel occlusion as measured by ferric chloride injury.
Materials and methods
Antibodies and reagents
All reagents were purchased from Thermo Fisher Scientific unless otherwise stated. Chronolume, used for the detection of secreted ATP, was purchased from Chrono-log corporation (Havertown, PA). Anti-pSyk Y525/526 (mouse Y519/520), anti-pLAT (Y220), anti-LAT, anti-pSyk S297 (mouse S291), anti-pPLCγ2 Y1217 and anti-Akt S473 were purchased from Cell Signaling Technology (Beverly, MA). Anti-pSyk Y352 (Y346 in mice) and anti-pSyk Y348 (Y342 in mice) were purchased from Abcam (Cambridge, MA). ERK and pERK were purchased from Sigma (Burlington, MA). Anti-Syk [Syk(SYK-01) cat# sc-51 703; mouse monoclonal antibody raised against aa 5–360] and anti- PLCγ2 [PLCγ2(B-10) cat# sc-5283; mouse monoclonal antibody raised against aa 626–985] were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The absence of SykS in the Syk (S291A) platelets was confirmed by probing with a Syk rabbit polyclonal antibody from Cell Signaling (cat# 2712) which was raised against a peptide surrounding aa 628 in the c-terminal and Syk (cat#31398) which was raised against a peptide surrounding Asn463. Two-color analysis of mouse platelet activation was purchased from Emfret Analytics (Eibelstadt, Germany). Odyssey blocking buffer and secondary antibodies IRDye 800CW goat anti-rabbit and IRDye 680LT goat anti-mouse were purchased from Li-Cor (Lincoln, NE). CRP-XL was purchased from Dr Richard Farndale at the University of Cambridge. AYPGKF was purchased from GenScript (Piscataway, NJ).
Animal housing and production
Mice were housed in a pathogen-free facility, and all animal procedures were approved by the Temple University Institutional Animal Care and Use Committee (protocol #4864). Syk S291A mice were produced in the C57BL/6 background by Taconic/Cyagen (Santa Clara, CA) on a fee for service basis.
Preparation of mouse platelets
Mouse blood was collected, and platelets were isolated as previously described.Citation26 The resulting platelets were counted using a Hemavet 950FS blood cell analyzer (Drew Scientific, Dallas, TX). Platelet counts were adjusted to a final concentration of 1.5 × 108 cells/mL in N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid-buffered (pH 7.4) Tyrode’s solution containing 0.2 U/mL apyrase.
Platelet aggregation and ATP secretion
All platelet aggregation and secretion experiments were carried out using a lumi-aggregometer (Chrono-log) at 37°C under stirring conditions. Platelet aggregation was measured using light transmission, and ATP secretion was measured using Chrono-lume (a luciferin/luciferase assay).
Western blotting
Western blotting procedures were performed as described previously.Citation26 Briefly, platelets were stimulated for the indicated time points in a Lumi-aggregometer with a GPVI agonist. The reaction was stopped by precipitating the platelet proteins using 0.6 N HClO4 and washed with water prior to the addition of sample loading buffer. Platelet protein samples were then boiled for 5-min prior to resolution by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were then blocked using Odyssey blocking buffer and incubated overnight with primary antibodies against the indicated protein. The membranes were then washed with tris-buffered saline containing 0.1% Tween-20 prior to incubation with appropriate secondary antibodies for 1 h at room temperature. The membranes were washed again and imaged using a Li-Cor Odyssey infrared imaging system.
Tail bleeding assay
Mouse tail bleeding was conducted as previously described.Citation27 Mice aged 4–6 weeks were anesthetized prior to the amputation of the distal 3 mm of the tail. The tail was then immersed in 37°C saline, and bleeding was monitored. If bleeding continued for greater than 600 s, then bleeding was halted manually by applying pressure.
Flow cytometry
Surface expression of P-selectin was determined as previously described.Citation28 Briefly, aliquots of isolated murine platelets were preincubated with the indicated concentrations of agonist for 5 min at 37°C. An aliquot containing 106 platelets was gently mixed with 5 ul FITC-anti-p-selectin (Emfret) and incubated for 15 min at RT. Platelets were fixed with 1% paraformaldehyde in PBS. For the surface expression of GPVI (clone JAQ1; Emfret) and αIIb (CD41), aliquots of isolated murine platelets were incubated with 10 µl of the corresponding antibody for 15 min at RT and treated as above. All determinations were performed on a FACSymphony flow cytometer.
Carotid artery injury
FeCl3 was used to injure the carotid artery as previously described.Citation27 Mice aged 10–12 weeks were anesthetized, and the carotid artery was exposed. A baseline blood flow reading was obtained using a Transonic T402 flowmeter (Ithaca, NY). The carotid artery was injured using a 1 × 1 mm piece of filter paper saturated with 7.5% FeCl3 for 90 s. The filter paper was removed, and blood flow was recorded.
Statistics
All statistical analysis was performed using Microsoft Excel, and data were analyzed using two-way ANOVA. All the data are presented as means ± SD of at least three independent experiments.
Results
Generation and characterization of Syk (S291A) knock-in mice
Studies in several cell lines,Citation21,Citation22 as well as human platelets,Citation25 indicated the importance of S291 in Syk regulation. However, studies using inhibitors are difficult to interpret the impact of S291 phosphorylation on the function. To determine the role of this phosphorylation in ITAM-receptor signaling, we generated a Syk (S291A) knock-in mouse using the CRISPR/Cas9 technique through a commercial source (Taconic/Cyagen, Santa Clara, CA). The coding sequence for S291 (TCC) was mutated to Ala (GCG), resulting in a mutant sequence of ACGCGT recognition site for a restriction enzyme MluI. The Syk S291A mutation was confirmed by both digesting the PCR product with a restriction enzyme, MluI, and by sequencing. The oligonucleotides used for PCR are forward primer 5′- CATGCAGGAAATCTCCCTGGG and reverse primer 5′-TGTAAAGGGCAAGGCAACGTGG. MluI cannot cleave the WT nucleotide sequence (ACTCCT) but can cleave the knock-in sequence (ACGCGT). Therefore, the results after the digest are as follows: WT (+/+) 362 bp product, heterozygous (±) 362, 213 and 149 bp products, and homozygous (-/-) 213 and 149 bp products (). The faint band seen in the homozygous knock-in at 362 is very likely due to incomplete digestion by the restriction enzyme. We verified the genotyping by sequencing the PCR product (). We also sequenced total Syk cDNA to ensure that there were no unwanted mutations. Both heterozygous Syk S291A knock-in mice and homozygous Syk S291A knock-in mice bred normally and produced pups at expected Mendelian ratios. Blood cell counts were not altered in Syk S291A knock-in mice ().
Figure 1. Generation of Syk S291A knock-in mice. (A) Restriction enzyme digest of PCR products from Syk S291A homozygous knock-in (SykS291A/S291A), Syk S291A heterozygous (SykS291A/WT), and WT littermate control (SykWT/WT) DNA. The oligonucleotides used for PCR are forward primer 5’- CATGCAGGAAATCTCCCTGGG and reverse primer 5’-TGTAAAGGGCAAGGCAACGTGG. Mutation of S291 to Ala, creates a site for MluI. WT PCR product is 362 bp while that of knock in is 213 and 149 bp. (B) Sequence analysis of PCR products of wild-type (WT) and S291A homozygous mouse DNA. (C) Repre-sentative Western blot (using 8% SDS-PAGE) showing that Syk S291 phosphorylation is present in CRP-stimulated WT platelets but absent in Syk S91A knock-in platelets. UN = Unstimulated sample.
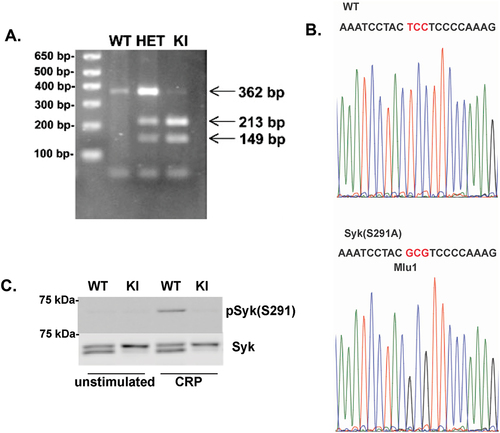
Table I. Blood cell counts from Syk S291A and WT littermate control mice. WBC = white blood cell, NE = neutrophil, LY = lymphocyte, RBC = red blood cell, PLT = platelet, MpV = mean platelet volume. *p < .05 compared to WT, n = 12.
To also confirm the S291A mutation at the protein level, we isolated platelets from Syk S291A and WT littermate control mice and stimulated them with a GPVI agonist, collagen-related peptide (CRP). We performed Western blot analysis to visualize phosphorylation of S291 and observed a robust band in WT mouse platelets treated with CRP, but no band at the expected molecular weight in samples obtained from Syk S291A mouse platelets ().
Inadvertent generation of SykS knockout mouse
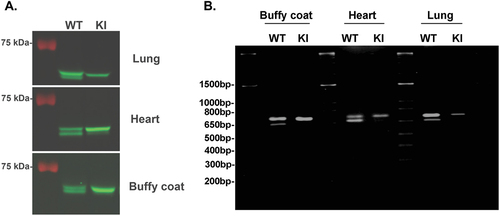
As can be seen in , the total Syk panels demonstrate only the long form of Syk (SykL) in the S291A knock in mice, while the wild-type (WT) counterparts show both the SykL and SykS isoforms. We were puzzled by this observation and investigated whether this phenomenon is selective for platelets or occurs in other tissues as well. As shown in , cellular extracts from the lung, heart, and the buffy coat of blood (comprising leukocytes) show both isoforms in WT mice, but only SykL in the S291A knock in mice. These results indicate that the insertion of the mutation in the exon 7 of Syk resulted in the disappearance of SykS protein in general. This could be due to the generation of a cryptic alternative splice site resulting in a very short form of Syk comprising only products of exons 1–6, or rapid destruction of this protein. We attempted to detect the very short form of Syk which should be about 30 kDa, but no such protein was detected (supplemental Figure S1). We also tried to detect the mRNA for both SykL and SykS isoforms in these tissues using RTPCR. The primers used were 5’-CTGAAGGAGAACCTCATCAGGG-3’ (corresponding to 841–863) as sense primer and 5’-GGCTCCTGTCCAGGTAGACC-3’ (corresponding to 1463–1442) as the antisense primer. As can be seen in , both isoforms of Syk were detected in the lung, heart, and buffy coat from WT mice but only SykL product in these tissues from Syk S291A knock in mice. No smaller RT-PCR product was seen in these tissues, indicating that alternate splicing resulting in smaller mRNA did not occur in these tissues. These data also indicate that the mRNA for the SykS isoform probably did not form in these tissues. Thus, the Syk S291A mice have acquired a selective knock out of the SykS isoform mice, which might be helpful for investigating the role of Syk B in other systems.
Figure 2. Analysis of Syk isoforms in multiple tissues of Syk S291A knock-in mouse: A) Cellular extracts from different tissues from Syk S291A mice (as indicated) and wild-type littermates were run on 8% SDS-PAGE and analyzed by western blot using total Syk antibody. Two isoforms are seen in wild-type tissue extracts but only long form of Syk is seen in the tissue extracts from the Syk S291A knock in mice. B) RNA was isolated from different tissues from Syk S291A mice and wild-type litter mates was analyzed by RTPCR. The primers used were 5’-CTGAAGGAGAACCTCATCAGGG-3’ (corresponding to 841–863) as sense primer and 5’- GGCTCCTGTCCAGGTAGACC-3’ (corresponding to 1463–1442) as the antisense primer.
Syk S291A platelets show enhanced GPVI-mediated aggregation and secretion
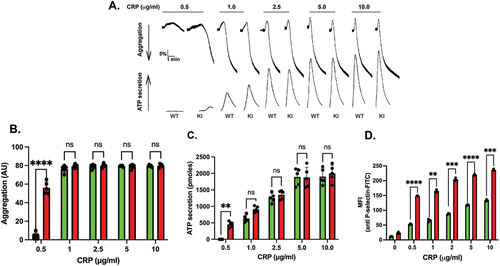
Platelets from Syk S291A mice respond normally to the PAR-4 agonist AYPGKF () or the purinergic agonist 2-MeSADP (). This result is consistent with the lack of involvement of Syk in signaling through these platelet receptors. Since Syk activity is crucial to platelet activation by GPVI agonists, we evaluated the effect of Syk S291A on GPVI-mediated aggregation and secretion by activating platelets isolated from Syk S291A and WT littermate control mice with varying concentrations of CRP. Our results indicated that there was very little aggregation () or ATP secretion () in WT platelets at very low concentrations of CRP, whereas Syk S291A platelets aggregated and secreted under these conditions (). This enhanced response was not observed when a high concentration of CRP was used (). Consistently, P-selectin surface expression was enhanced in the platelets from S291A knock in mice compared to WT littermates upon stimulation with varying concentrations of CRP ().
Figure 3. GPCR- and ADP-mediated platelet reactivity is intact in Syk S291A mice. (A) Representative aggregation and secretion tracings of platelets from WT and Syk S291A mice activated with the PAR4 agonist AYPGKF. (B) Quantification of ATP secretion of platelets from Syk S291A (red bars) and WT littermate control mice (green bars) stimulated with various concentrations AYPGKF. (C) Representative aggregation and secretion tracings of platelets from WT and Syk S291A mice stimulated with the P2Y agonist 2-MeSADP. (D) Quantitation of ATP secretion of platelets from Syk S291A (red bars) and WT mice (green bars) stimulated with various concentrations of 2-MeSADP. n = 3.
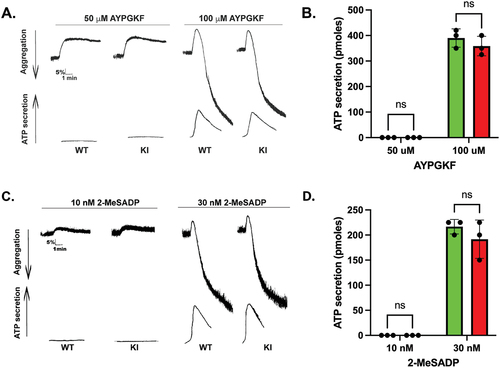
Figure 4. GPVI-mediated aggregation and secretion are potentiated in Syk S291A platelets. (A) Representative aggregation and secretion tracings of Syk S291A (red bars) and WT littermate control (green bars) platelets stimulated with the indicated concentrations of CRP, under conditions of aggregation in the absence of any feedback inhibitors. (B) Quantification of aggregation, (C) ATP secretion and (D) p-Selectin surface expression from multiple independent experiments. p. value ****<0.0001, n = 5.
GPVI-mediated signaling is enhanced in Syk S291A platelets
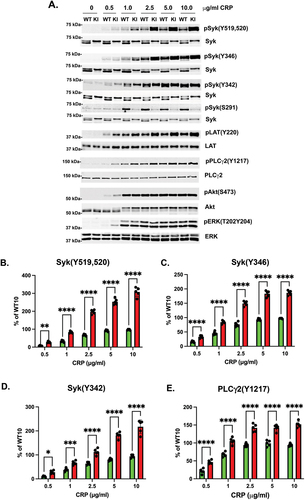
Syk is an essential component of the signaling pathway downstream from ITAM-bearing receptors. Therefore, we analyzed the phosphorylation of key proteins involved in the signaling cascade downstream of GPVI stimulation. After activation with low concentrations of CRP, under the conditions of little or no aggregation or secretion, there was little phosphorylation of Syk Y519/520, Syk Y342, LAT Y191, or PLCγ2 Y1217 in WT platelets, but phosphorylation of all these sites was enhanced in Syk S291A platelets (). Contrary to the aggregation and secretion data, the phosphorylation of Syk Y519/520, Syk Y342, LAT Y191, or PLCγ2 Y1217 was enhanced at all concentrations of CRP tested (). ERK and Akt phosphorylation were also enhanced when Syk S291A platelets were activated with CRP compared to the WT littermates (). These data collectively indicate that signaling downstream of GPVI is enhanced in Syk S291A platelets.
Figure 5. CRP-mediated signaling is increased in Syk S291A platelets. (A) Representative Western blots showing the indicated that phosphorylated and total protein in Syk S291A and WT littermate control platelets stimulated with the indicated concentrations of CRP for 3 minutes under aggregating conditions. (B-H) Quantification of the indicated phosphorylated residue expressed as a percentage of the maximum WT (WT10). p value****<0.0001, n = 5.
We have previously shown that platelets and B cells do not express ZAP-70.Citation29 This rules out the possibility of ZAP-70 mediating the effects in platelets. We have further evaluated the surface levels of GPVI on platelets from WT and Syk S291A mutant mice using flow cytometry. The results, shown in supplemental Figure S2S, indicate that there is no difference in the surface level of GPVI between the WT and S291A mouse platelets. Hence, the negative regulatory effect seen in S291A mice is not due to an enhanced surface expression of GPVI receptors.
Syk S291A is differentially involved in hemostasis and in vivo thrombus formation
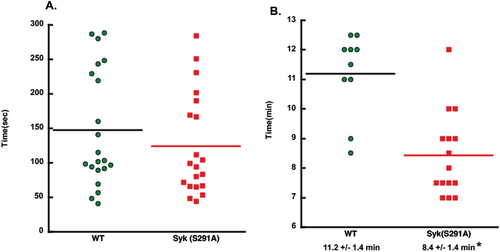
To determine the impact of Syk S291A on hemostasis, we performed tail bleeding assay on litter-matched WT and homozygous Syk S291A mice. We found no significant differences in the bleeding times (). These data suggest that Syk S291 phosphorylation exerts no effect on in-vivo platelet-dependent hemostasis. To determine whether thrombus formation is impacted by the impaired phosphorylation of S291 on Syk, we injured the carotid artery in Syk S291A and WT littermate control mice with 7.5% FeCl3 for 90 s and monitored its occlusion. Syk S291A mice formed a stable occlusion in statistically less time than WT mice (). These data argue that the positive effect of the Syk S291A mutation we observed for GPVI-mediated signaling and platelet responses in vitro differentially translates to more rapid physiological responses in vivo.
Figure 6. In vivo thrombus formation is enhanced in Syk S291A knock-in mice. (A) Scatter plot showing the time it took for bleeding to stop during tail bleeding experiments conducted on homozygous SykS291A/S291A knock-in and WT (SykWT/WT) littermate control mice 4–6 weeks of age in a blind fashion. (B) Scatter plot of the time to occlusion in WT and Syk S291A knock-in mice in a blind fashion following 7.5% FeCl3 injury on the carotid artery and analyzed by student’s test.
Discussion
It has been known that Syk is phosphorylated on multiple tyrosine residues thereby regulating Syk activity and Syk-mediated signaling.Citation1,Citation11,Citation30–32 Syk is also predominantly phosphorylated on Ser 297 with implications in binding to several signaling molecules. This phosphorylation was mainly studied in B lymphocytes and platelets and was found to be dependent on Akt or PKC. As these studies were performed with pharmacological inhibitors, with nonselective inhibitory effects on other kinases, it is not clear which kinase phosphorylates Syk 297. In addition, the functional effects of S297 phosphorylation in these cells are complicated to interpret due to the nonselective effects of the inhibitors. With a goal to evaluate the functional role and signaling effects of human Syk S297 (mouse 291) phosphorylation, we generated a knock-in mouse Syk S291A and evaluated its effect on signaling and function in platelets.
The Syk S291A mutation does not seem to affect the breeding or growth of the mice, and the blood cell counts were comparable to WT littermates. Quite interestingly, the short form of Syk, SykS, disappeared from several tissues tested, indicating that we inadvertently created a SykS knockout mouse (of course with a mutant SykL). The reason for this deletion of SykS is unclear, but most likely due to the creation of a cryptic alternative splice site that resulted in degradation of the abnormal protein generated. To the best of our knowledge, it is nearly impossible to generate SykS knockout mouse without also deleting SykL. Hence, our mouse provides a unique reagent to study the role of SykS and the role of SykL (with a mutation) without interference from SykS, in some systems. Both SykL and SykS have been attributed to diverse functions, particularly in cancer cells.Citation13
Our studies demonstrate that Syk S291A platelets show an increased downstream signaling as well as platelet function downstream of GPVI. These mice have also shown increased thrombotic tendency when tested in a ferric chloride model. These data indicate that phosphorylation of Syk on S291 negatively regulates its function. It is quite possible that SykS competes with SykL, and lack of SykS removed this competition and resulted in increased signaling by SykL. We believe that this is unlikely due to studies with pharmacological inhibitors where both isoforms are present but inhibition of S291 phosphorylation correlated with increased signaling events. While the studies with pharmacological inhibitors were correlative in nature, the studies with knock-in mice are more definitive.
In mice, both B cells and platelets express SykL and SykS but not Zap70.Citation29 Zap70 is a homolog of Syk expressed in T cells and is more like Syk S as it lacks the S291 equivalent motif.Citation33 However, Zap70 functions normally in the activation of PLCγ1, despite lacking the S291 motif. Thus, we can presume that SykS also functions normally in B cells and platelets in the activation of PLCγ2. However, we can only demonstrate it by generating Syk exon 7 deficient mice and evaluating the platelets.
Signaling pathways in platelets are quite complex and are often influenced by feedback signaling. When platelets are activated, they release the contents of their granules, which contain several platelet agonists. They also generate thromboxane A2, another platelet agonist. These released and generated platelet agonists act on platelet receptors to trigger signaling cascades that influence the platelet signaling and function. Interestingly, pan-PKC inhibitors are used to inhibit platelet secretion and hence they not only interfere with the primary signaling but also block feedback signaling by secreted agonists. On the contrary, pan-PKC inhibitors enhance the generation of thromboxane A2,Citation34 thereby enhancing certain other pathways. Thus, the interpretation of the effects of a PKC inhibitor becomes complicated as to whether it is due to the inhibition at the primary level or the feedback level. Furthermore, using enzyme inhibitors, Mohammad et al.Citation23 showed that Akt (PKB) is the enzyme responsible for phosphorylating Syk 297 in B lymphocytes. Opposing this view, Makhoul et al.Citation25 demonstrated that PKC is the enzyme that phosphorylates Syk 297 in human platelets. Subsequent to the submission of our manuscript, Jurk and coworkers identified PKC as the S/T kinase responsible for Syk S297 phosphorylation.Citation35 In platelets, Akt is activated by ADP which is secreted from the granules upon platelet activation, and PKC inhibitors block this secretion of ADP and hence Akt activation.Citation36 Thus, it is quite possible that the effects seen with the PKC inhibitors are due to inhibition of secretion and consequent inhibition of Akt activation, in platelets. In addition, we will not know whether PKC is activating an S/T kinase that is responsible for S291 phosphorylation or PKC directly phosphorylates S291. Therefore, we cannot conclusively ascertain which S/T kinase phosphorylates Syk Ser 297.
We have previously shown that PKC inhibitors negatively regulate Syk phosphorylation and activity in human platelets but not in murine platelets.Citation24 It is possible that the PKC inhibitors block secretion from platelets and thus they block the negative feedback of secreted components on Syk activation. Thus, PKC inhibitors could elicit enhanced Syk phosphorylation and activity in human platelets. However, we did not observe such a phenomenon in murine platelets. This leads to the possibility that human platelets, but not murine platelets, contain certain components that regulate Syk phosphorylation which could be blocked by PKC inhibitors. Our data are consistent with the studies of Mohammed et al.,Citation23 who have shown that phospho serine mimicking Syk S291D mutant reduces Syk phosphorylation. Further studies using the Syk S291D knock in mice might confirm the significance of S291 phosphorylation in the regulation of Syk tyrosine phosphorylation and downstream signaling events. The possibilities of PKC regulatable components in human but not murine platelets, the unknown nature of the ST kinase phosphorylating S291, and the nonselective nature of PKC inhibitors on other kinases make the system quite complex and limit our ability to provide definitive conclusions.
Upon GPVI stimulation, Src family kinases phosphorylate the ITAM; this helps bind Syk to it and get activated.Citation37 Subsequently, Tec kinases and PLCγ2 are activated, downstream of which PKC isoforms are activated.Citation37 Hence, in this cascade, PKC appears to be the first Ser/Thr kinase that is activated, which also activates Akt through secretion.Citation36 It is possible that the feedback loop is what causes regulation of Syk through phosphorylation of Ser 297. It is also possible that a Ser/Thr kinase, yet to be identified, directly phosphorylates S297 upon activation (unfolding) of Syk when it binds to phospho-ITAM. These possibilities require further investigation.
In summary, we conclude that Ser 291 phosphorylation on Syk negatively regulates its downstream-signaling events, and mutation of this site results in both enhanced signaling and platelet functional responses mediated by GPVI.
Authors statement
All authors have significantly contributed to the article and have read and approved the manuscript.
Supplemental Material
Download PDF (77.3 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data concerning this report are available in the manuscript.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/09537104.2024.2369766.
Additional information
Funding
References
- Geahlen RL. Syk and pTyr’d: signaling through the B cell antigen receptor. Biochim Biophys Acta. 2009;1793(7):1115–9. doi:10.1016/j.bbamcr.2009.03.004.
- Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10(6):387–402. doi:10.1038/nri2765.
- Bradshaw JM. The Src, Syk, and Tec family kinases: distinct types of molecular switches. Cell Signal. 2010;22(8):1175–84. doi:10.1016/j.cellsig.2010.03.001.
- Tsang E, Giannetti AM, Shaw D, Dinh M, Tse JK, Gandhi S, Ho H, Wang S, Papp E, Bradshaw JM. Molecular mechanism of the Syk activation switch. J Biol Chem. 2008;283(47):32650–9. doi:10.1074/jbc.M806340200.
- Rowley RB, Bolen JB, Fargnoli J. Molecular cloning of rodent p72Syk. Evidence of alternative mRNA splicing. J Biol Chem. 1995;270(21):12659–64. doi:10.1074/jbc.270.21.12659.
- Yamashita T, Kairiyama L, Araki M, Nagasawa S. Evidence for involvement of two isoforms of Syk protein-tyrosine kinase in signal transduction through the high affinity IgE receptor on rat basophilic leukemia cells. J Biochem (Tokyo). 1998;123(6):1199–207. doi:10.1093/oxfordjournals.jbchem.a022061.
- Duta F, Ulanova M, Seidel D, Puttagunta L, Musat-Marcu S, Harrod KS, Schreiber AD, Steinhoff U, Befus AD. Differential expression of spleen tyrosine kinase Syk isoforms in tissues: effects of the microbial flora. Histochem Cell Biol. 2006;126(4):495–505. doi:10.1007/s00418-006-0188-z.
- Wang L, Duke L, Zhang PS, Arlinghaus RB, Symmans WF, Sahin A, Mendez R, Dai JL. Alternative splicing disrupts a nuclear localization signal in spleen tyrosine kinase that is required for invasion suppression in breast cancer. Cancer Res. 2003;63(15):4724–30.
- Kumar Singh P, Dangelmaier CA, Reddy Vari H, Tsygankov AY, Kunapuli SP. Biochemical characterization of spleen tyrosine kinase (SYK) isoforms in platelets. Platelets. 2023;34(1):2249549. doi:10.1080/09537104.2023.2249549.
- Yanagi S, Inatome R, Takano T, Yamamura H. Syk expression and novel function in a wide variety of tissues. Biochem Biophys Res Commun. 2001;288(3):495–8. doi:10.1006/bbrc.2001.5788.
- Tsygankov AY. Non-receptor protein tyrosine kinases. Front Biosci. 2003;8(6):s595–635. doi:10.2741/1106.
- Coopman PJ, Mueller SC. The Syk tyrosine kinase: a new negative regulator in tumor growth and progression. Cancer Lett. 2006;241(2):159–73. doi:10.1016/j.canlet.2005.11.004.
- Hong J, Yuan Y, Wang J, Liao Y, Zou R, Zhu C, Li B, Liang Y, Huang P, Wang Z, et al. Expression of variant isoforms of the tyrosine kinase SYK determines the prognosis of hepatocellular carcinoma. Cancer Res. 2014;74(6):1845–56. doi:10.1158/0008-5472.CAN-13-2104.
- Coebergh van den Braak RRJ, Sieuwerts AM, Kandimalla R, Lalmahomed ZS, Bril SI, van Galen A, Smid M, Biermann K, van Krieken J, Kloosterman WP, et al. High mRNA expression of splice variant SYK short correlates with hepatic disease progression in chemonaive lymph node negative colon cancer patients. PLOS ONE. 2017;12(9):e0185607. doi:10.1371/journal.pone.0185607.
- Prinos P, Garneau D, Lucier JF, Gendron D, Couture S, Boivin M, Brosseau JP, Lapointe E, Thibault P, Durand M, et al. Alternative splicing of SYK regulates mitosis and cell survival. Nature Structural & Molecular Biology. 2011;18(6):673–9. doi:10.1038/nsmb.2040.
- Ni B, Hu J, Chen D, Li L, Chen D, Wang J, Wang L. Alternative splicing of spleen tyrosine kinase differentially regulates colorectal cancer progression. Oncol Lett. 2016;12(3):1737–44. doi:10.3892/ol.2016.4858.
- Denis V, Cassagnard N, Del Rio M, Cornillot E, Bec N, Larroque C, Jeanson L, Jarlier M, Combes E, Robert B, et al. Targeting the splicing isoforms of spleen tyrosine kinase affects the viability of colorectal cancer cells. PLOS ONE. 2022;17(9):e0274390. doi:10.1371/journal.pone.0274390.
- Siraganian RP, de Castro RO, Barbu EA, Zhang J. Mast cell signaling: the role of protein tyrosine kinase Syk, its activation and screening methods for new pathway participants. FEBS Lett. 2010;584(24):4933–40. doi:10.1016/j.febslet.2010.08.006.
- Sada K, Takano T, Yanagi S, Yamamura H. Structure and function of Syk protein-tyrosine kinase. J Biochem. 2001;130(2):177–86. doi:10.1093/oxfordjournals.jbchem.a002970.
- Kulathu Y, Grothe G, Reth M. Autoinhibition and adapter function of Syk. Immunol Rev. 2009;232(1):286–99. doi:10.1111/j.1600-065X.2009.00837.x.
- Paris LL, Hu J, Galan J, Ong SS, Martin VA, Ma H, Tao WA, Harrison ML, Geahlen RL. Regulation of Syk by phosphorylation on serine in the linker insert. J Biol Chem. 2010;285(51):39844–54. doi:10.1074/jbc.M110.164509.
- Bohnenberger H, Oellerich T, Engelke M, Hsiao HH, Urlaub H, Wienands J. Complex phosphorylation dynamics control the composition of the Syk interactome in B cells. Eur J Immunol. 2011;41(6):1550–62. doi:10.1002/eji.201041326.
- Mohammad DK, Nore BF, Gustafsson MO, Mohamed AJ, Smith CIE. Protein kinase B (AKT) regulates SYK activity and shuttling through 14-3-3 and importin 7. Int J Biochem Cell Biol. 2016;78:63–74. doi:10.1016/j.biocel.2016.06.024.
- Buitrago L, Bhavanasi D, Dangelmaier C, Manne BK, Badolia R, Borgognone A, Tsygankov AY, McKenzie SE, Kunapuli SP. Tyrosine phosphorylation on spleen tyrosine kinase (Syk) is differentially regulated in human and murine platelets by protein kinase C isoforms. Journal of Biological Chemistry. 2013;288(40):29160–9. doi:10.1074/jbc.M113.464107.
- Makhoul S, Dorschel S, Gambaryan S, Walter U, Jurk K. Feedback regulation of Syk by protein Kinase C in human platelets. Int J Mol Sci. 2019;21(1):176. doi:10.3390/ijms21010176.
- Kostyak JC, Mauri BR, Dangelmaier C, Patel A, Zhou Y, Eble JA, Tsygankov AY, McKenzie SE, Kunapuli SP. TULA-2 deficiency enhances platelet functional responses to CLEC-2 agonists. TH Open. 2018;2(4):411–9. doi:10.1055/s-0038-1676358.
- Patel A, Kostyak J, Dangelmaier C, Badolia R, Bhavanasi D, Aslan JE, Merali S, Kim S, Eble JA, Goldfinger L, et al. ELMO1 deficiency enhances platelet function. Blood Adv. 2019;3(4):575–87. doi:10.1182/bloodadvances.2018016444.
- Daniel JL, Dangelmaier CA, Mada S, Buitrago L, Jin J, Langdon WY, Tsygankov AY, Kunapuli SP, Sanjay A. Cbl-b is a novel physiologic regulator of glycoprotein VI-dependent platelet activation. J Biol Chem. 2010;285(23):17282–91. doi:10.1074/jbc.M109.080200.
- Dangelmaier CA, Patchin M, Vajipayajula DN, Vari HR, Singh PK, Wright MN, Kostyak JC, Tsygankov AY, Kunapuli SP. Phosphorylation of spleen tyrosine kinase at Y346 negatively regulates ITAM-mediated signaling and function in platelets. J Biol Chem. 2023;299(7):104865. doi:10.1016/j.jbc.2023.104865.
- Siraganian RP, Zhang J, Suzuki K, Sada K. Protein tyrosine kinase Syk in mast cell signaling. Mol Immunol. 2002;38(16–18):1229–33. doi:10.1016/S0161-5890(02)00068-8.
- Sada K, Takano T, Yanagi S, Yamamura H. Structure and function of syk protein-tyrosine kinase. J Biochem (Tokyo). 2001;130(2):177–86. doi:10.1093/oxfordjournals.jbchem.a002970.
- Turner M, Schweighoffer E, Colucci F, Di Santo JP, Tybulewicz VL. Tyrosine kinase SYK: essential functions for immunoreceptor signalling. Immunol Today. 2000;21(3):148–54. doi:10.1016/S0167-5699(99)01574-1.
- Swamy M. ZAP70 holds the key to kinetic proofreading for TCR ligand discrimination. Nat Immunol. 2022;23(9):1293–4. doi:10.1038/s41590-022-01297-w.
- Garcia A, Shankar H, Murugappan S, Kim S, Kunapuli SP. Regulation and functional consequences of ADP receptor-mediated Erk2 activation in platelets. Biochem J. 2007;404(2):299–308. doi:10.1042/BJ20061584.
- Zhang P, Ungern-Sternberg S, Hastenplug L, Solari FA, Sickmann A, Kuijpers MJE, Heemskerk JWM, Walter U, Jurk K. Multi-phased kinetics and interaction of protein kinase signaling in glycoprotein VI-Induced platelet αIIbβ3 integrin activation and degranulation. Thromb Haemost. 2024; In press. 10.1055/a-2311-0117.
- Kim S, Foster C, Lecchi A, Quinton TM, Prosser DM, Jin J, Cattaneo M, Kunapuli SP. Protease-activated receptors 1 and 4 do not stimulate G(i) signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of G(i) signaling. Blood. 2002;99(10):3629–36. doi:10.1182/blood.V99.10.3629.
- Watson SP, Auger JM, McCarty OJ, Pearce AC. GPVI and integrin alphaIib beta3 signaling in platelets. J Thromb Haemost. 2005;3(8):1752–62. doi:10.1111/j.1538-7836.2005.01429.x.