
Abstract
Hemolysis is associated with thrombosis and vascular dysfunction, which are the pathological components of many diseases. Hemolytic products, including hemoglobin and hemin, activate platelets (PLT). Despite its activation, the effect of hemolysis on platelet clearance remains unclear, It is critical to maintain a normal platelet count and ensure that circulating platelets are functionally viable. In this study, we used hemin, a degradation product of hemoglobin, as a potent agonist to treat platelets and simulate changes in vivo in mice. Hemin treatment induced activation and morphological changes in platelets, including an increase in intracellular Ca2+ levels, phosphatidylserine (PS) exposure, and cytoskeletal rearrangement. Fewer hemin-treated platelets were cleared by macrophages in the liver after transfusion than untreated platelets. Hemin bound to glycoprotein Ibα (GPIbα), the surface receptor in hemin-induced platelet activation and aggregation. Furthermore, hemin decreased GPIbα desialylation, as evidenced by reduced Ricinus communis agglutinin I (RCA- I) binding, which likely extended the lifetime of such platelets in vivo. These data provided new insight into the mechanisms of GPIbα-mediated platelet activation and clearance in hemolytic disease.
Plain Language Summary
What is the context?
Hemolysis is a primary hematological disease. Hemolysis is a pathological complication of several diseases.
Hemin, a degradation product of cell-free hemoglobin, has been proven to be a more potent agonist than hemoglobin for directly activating platelets.
Platelet membrane glycoproteins (GP), including GPIb-IX and GPIIb/IIIa complexes, play crucial roles in platelet hemostasis.
Desialylation (loss of sialic acid residues) of GPIbα, is believed to regulate physiological platelet clearance through liver macrophages and hepatocytes.
What is new?
In this study, we evaluated the effects of hemolysis on platelet clearance. We first analyzed the influence of hemin at 0-50 μM on platelets in vitro before exploring the mechanism underlying hemin-induced platelet activation and its role in platelet clearance in vitro and in vivo.
Our analyses suggest that:
Hemin bound to GPIbα on the platelet surface with high affinity.
Platelet clearance occurred slowly in the liver and spleen after hemin treatment.
Platelets exhibited significant significantly reduced GPIbα surface expression and desialylation after hemin treatment.
Platelets exhibited significant significantly reduced GPIbα surface expression and desialylation after hemin treatment.
What is the impact?
This study provides new insights into the role of hemin in the mechanisms of GPIbα-mediated platelets activation and clearance in diseases associated with hemolysis.
Keywords:
Introduction
Hemolytic disease is a condition in which the lifespan of red blood cells is reduced. Several hematologic diseases, including paroxysmal nocturnal hemoglobinuria (PNH), thalassemia, and glucose-6-phosphate dehydrogenase deficiency, are primarily characterized by hemolysis. Hemolysis is also a pathological complication of diseases such as malaria,Citation1 sepsisCitation2 and sickle cell disease,Citation3 and is accompanied by platelet activation, hypercoagulability, thrombocytopenia, platelet-leukocyte aggregates and inflammation. Hemolysis is associated with thrombosis and vascular dysfunction. Hemolytic products, including hemoglobin and hemin, could activate platelets (PLT) directly. Meanwhile, a broad range of plasma Hb concentrations (0.37–6 μM) has been demonstrated to be directly correlated with the occurrence and severity of intravascular thrombosis and thromboembolism in PNH patients.Citation4 Hemin, a degradation product of cell-free hemoglobin, has been shown to be a more potent agonist than hemoglobin for directly activating platelets.Citation5 It is a signaling molecule that mediates various biochemical processes, such as inflammation, transcription, and signal transduction, via transient binding to various proteins.Citation6,Citation7 Annarapu GK. et al. showed that hemin-stimulated human platelet mitochondrial oxidant production induces targeted granule secretion.Citation8 Moreover, recently, hemin was shown to activate human and mice platelets via CLEC-2 and GPVI/FcRγ.Citation8
Platelets adhere to and are activated via ligand-receptor recognition and intracellular signaling pathways. Platelet adhesion, activation, and aggregation are crucial for hemostasis and thrombosis. Platelet membrane glycoproteins (GP), including GPIb-IX and GPIIb/IIIa complexes, play crucial roles in platelet hemostasis. The former is the second-most abundant platelet membrane glycoprotein.Citation9 GPIb is composed of subunits of GPIbα and GPIbβ that noncovalently combine with glycoprotein GPIX to form GPIb-IX.Citation10,Citation11 The extracellular domain binding of the von Willebrand factor is critical for initial platelet adhesion at the site of injury,Citation12 and triggers downstream signaling in platelets, including activation, aggregation and phosphatidylserine (PS) exposure.Citation13 The most heavily glycosylated platelet membrane surface protein is GPIbα.Citation14 The terminal glycan residue of GPIbα is sialic acid, linked to a penultimate β-galactose (β-gal) and antepenultimate GlcNAc. Desialylation (loss of sialic acid residues) of major platelet surface glycoproteins, primarily GPIbα, is believed to regulate physiological platelet clearance and lead to the clearance of human and mouse platelets by liver macrophages and hepatocytes.Citation15–19 Another mechanism that contributes to the clearance of old platelets from mice is apoptosis.Citation20
Despite its known effects on platelet activation, the role of hemin in platelet clearance remains unclear. We investigated the effect of hemin at increased labile (free and weakly bound) heme/hemin in plasma concentrations between 2 μM and 50 μMCitation21,Citation22 in patients with hemolytic diseases. Subsequently, we assessed the mechanism underlying hemin-induced platelet activation and its role in platelet clearance via in vitro and in vivo experiments. To the best of our knowledge, this is the first study to systematically investigate the interactions between hemin and platelets and is expected to help us understand the mechanism by which hemin regulates platelet clearance in hemolytic diseases.
Materials and methods
Animals
Male wild-type C57BL/6J mice (6–8 weeks) were purchased from Vital River (Beijing, China). C57BL/6-Tg (GFP) transgenic GFP and tdTomato mice were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). All animal studies were performed in accordance with the guidelines of the National Beijing Center for Drug Safety Evaluation and Research (IACUC-DWZX-2023-528).
Isolation of apheresis platelets (APs) from humans
APs were isolated from healthy human volunteers at the Chinese People’s Liberation Army (PLA) General Hospital (Beijing, China). This study was designed and conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the National Beijing Center for Drug Safety Evaluation and Research. All the donors met the criteria for AP donation in China. No patient-identifying information was involved in this study; therefore, informed consent was not required. The APs were washed twice with calcium-free Tyrode’s buffer [1 L Tyrode’s buffer contains 8.0 g NaCl, 0.2 g KCl, 2.383 g HEPES, 0.056 g NaH2PO4 *2 H2O, 0.203 g MgCl2 *6 H2O and 1.0 g glucose] (Yuanye Biotech, China), centrifuged at 800 × g for 15 min at 37°C, and then suspended in calcium-free Tyrode’s buffer.
Preparation of platelets from mice
Mice were anesthetized, and whole blood was collected via cardiac puncture using a 1-mL syringe containing 14% anticoagulant (CPDA-1). Leukoreduced blood samples were collected and pooled into sterile microcentrifuge tubes. Platelet-rich plasma (PRP) was prepared by gentle centrifugation (two rounds of 15 min each at 150 × g).
Preparation of hemin
A 50 mM hemin (from Porcine, ≥ 96.0% (HPLC), Sigma-Aldrich) stock solution was dissolved with 1.4 M sodium hydroxide and diluted to 50 μM working stock in modified calcium-free Tyrode’s buffer for all experiments.
Co-culture experiments
For platelet-hemin co-culture experiments, 5–10 × 106 platelet suspension was treated with six concentrations of hemin (0, 5, 10, 25, 40 and 50 μM) for 15 min at 37°C or pre-treated with a GPIbα blocking anti-GPIbα mAb (5 μg/mL; Biolegend, clone H1P1) in 1 mL Tyrode’s buffer for 30 min at 37°C before the cells were washed with Tyrode’s buffer. The platelets were then used for flow cytometry and morphological observation by microscopy.
Flow cytometry
Platelet activation was measured by staining with FITC-labeled anti-human CD62P (P-selectin) antibody (5 μg/mL) (Biolegend, Clone AK4) or PE-labeled anti-human CD42b (GPIbα) antibody (5 μg/mL, Biolegend, Clone H1P1) for human platelets. PS exposure was assessed using 5 μL annexin V-PE (BD Biosciences). Desialylation was detected using fluorescein-labeled RCA-1 (Vector Laboratories, USA) on human platelets. An ROS assay kit (Beyotime, China) was used to measure the total intracellular reactive oxygen species (ROS) production according to the manufacturer’s instructions. Mitochondrial membrane potential was determined using the JC-1 Assay Kit (Beyotime, China) according to the manufacturer’s instructions and quantified by flow cytometry. The fold increase was represented as a percentage increase from the gated control platelets set at 1% positivity. All isotype control antibodies were purchased from the same manufacturer as the detection antibodies.
Intracellular free-calcium measurements
For imaging intracellular Ca2+ ([Ca2+]i) transients induced by hemin treatment, human platelets were incubated with 2 μM Fluo-4 AM (Molecular Probes, DojinDo) for 30 min at 37°C. Cells were washed with HBSS, incubated for 20 min at 37°C in HBSS medium, and then stimulated with hemin. Images were captured immediately every 10 s using a confocal microscope (Zeiss, Germany). The recorded images were analyzed and quantified using Image J software (NIH).
For image analysis, background correction was performed by subtracting the intensity of untreated control platelets from that of hemin-treated platelets. F0 was calculated by subtracting the initial background from the fluorescence intensity. F indicates fluorescence intensity at each time point. ΔF/F0 = (F – F0)/F0, indicated the Ca2+ concentration change after hemin stimulation.
Confocal microscopy for studying cytoskeletal protein dynamics in hemin-treated platelets
To evaluate the cytoskeletal protein dynamics in hemin-treated platelets, human platelets were allowed to settle on collagen-coated coverslips for 1 h, followed by incubation, washing with PBS, fixing with 4% paraformaldehyde (Merck), and permeabilization with 0.1% Triton X-100 (Sigma-Aldrich). Finally, platelets were stained with FITC-conjugated phalloidin (Sigma-Aldrich) to analyze F-actin and subsequent incubation with anti-β-tubulin antibodies (Abcam, Clone EPR1330) followed by Cy3-labeled secondary antibodies. Finally, all the coverslips were washed and viewed under a confocal laser scanning microscope (Perkin Elmer, USA).
Light transmission aggregometry
Human PRP (1 × 109/mL; 250 μL) was incubated with concentration-dependent hemin (5–50 μM) for 15 min at 37°C or pre-treated with anti-GPIbα blocking antibody (H1P1) (20 ug/mL) for 30 min at 37°C before aggregation. Platelet aggregation was assessed by light transmission aggregometry using Agg RAMTM (Roche Diagnostics, UK) in response to TRAP-6 (1.5 μM). Platelet-poor plasma (PPP) represented 100% aggregation as the control, whereas hemin-treated PRP showed 0% aggregation before stimulation. The results are presented as % aggregation, representing the maximum amplitude after stimulation.
Platelet aggregometry by confocal microscopy
For the assessment of platelet aggregation by confocal microscopy, 10 µL human PRP (1 × 109/mL) was allowed to settle in a collagen-coated chamber for 30 min at 37°C, followed by incubation with different concentrations of hemin (5–50 μM) for 1 h at 37°C and washed by calcium-free Tyrode’s buffer. All samples were photographed using a confocal microscope (Zeiss) after the platelets adhered to the bottom of the chamber. The platelets that adhered to the collagen-coated pore walls were activated and aggregated.
Western blotting
Platelets were washed twice and lysed. Total proteins in the cell lysates were quantified using a BCA assay kit. The same amounts of protein aliquots were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and the resulting protein bans were transferred to PVDF membranes. The membranes were blocked with 4% bovine serum albumin in TBST and incubated with anti-human CD42b antibody (Abcam, Clone EPR6995) and anti-human GAPDH antibody (Abcam) at 4°C overnight. The membranes were washed and incubated with horseradish peroxidase-conjugated secondary antibodies and then developed using an ECL Kit (Sigma-Aldrich).
Scanning electron microscopy (SEM)
For SEM imaging, the platelets were washed and fixed in 2.5% glutaraldehyde for 24 h at 4°C and treated for gradual desiccation. The specimens were sputter-coated with 20 nm gold and used for SEM observation (Hitachi, Japan).
Transmission electron microscopy (TEM)
For TEM imaging, the platelets were washed and fixed in 2.5% glutaraldehyde overnight at 4°C and washed with PBS, followed by post-fixation with 1% osmium tetroxide on ice for 1 h. Samples were dehydrated using a graded ethanol series and embedded in Epon. The specimens were used for TEM observations (Hitachi, Japan).
Generation of mouse bone marrow-derived macrophages (BMDMs)
As BMDM were usually employed to represent Kupffer cell in the liver in the researches that focus on the clearance of blood cells that transfused, the phagocytosis experiment of platelets in vitro was conducted by BMDM.
Bone marrow monocytes prepared from the femurs and tibias of tdTomato mice were cultured at a density of 2 × 106 cells/mL in 6-well plates with 25 ng/mL murine macrophage colony-stimulating factor (Peprotech, Israel) supplemented with complete Dulbecco’s modified Eagle medium (DMEM) (Life Technologies Corporation, USA). The medium was replaced with 2 mL of fresh medium containing M-CSF on days 3 and 5. Adherent cells were used as BMDMs on day 6.
Platelet clearance in vivo
A novel model was established to monitor the lifespan of platelets in circulation after transfusion. Platelets from GFP+ transgenic mice were GFP-positive (GFP+) and distinguishable from wild-type (WT) recipient platelets by FACS. GFP+ murine platelets were incubated with hemin (0–50 μM) for 15 min at 37°C and washed. Subsequently, mice were intravenously injected with 2 × 108 platelets in 300 μL PBS. The control mice received the same volume of untreated GFP+ platelets under identical conditions. After transfusion of GFP+ platelets, whole-blood samples were obtained from mice anesthetized by retro orbital bleeding at 4, 24, and 48 h. Platelet circulation was measured by the enumeration of fluorescently labeled platelets in the blood. The 20-min recovery of GFP+ murine platelets in each mouse was set at 100%, that is 0% clearance.
Platelet clearance in vitro
Platelet clearance in vitro was quantified by measuring the levels of GFP+ murine platelets in tdTomato BMDMs using flow cytometry. Briefly, 1–5 × 107 GFP+ platelets treated with hemin (0–50 μM) alone were added to 2 × 106 tdTomato BMDMs in 2 mL of complete DMEM. In another setup, platelets were pre-treated with a specific blocking antibody to GPIbα (20 µg/mL) for 30 min before being added. After 4 h of phagocytosis, BMDMs were washed for removal of noningested platelets and detected by FACS.
Immunohistochemistry
After the in vivo platelet clearance studies, the mice were euthanized, and their livers and spleens were harvested at 48 h and snap-frozen in liquid nitrogen. The frozen tissues were sectioned (5-mm-thick) and fixed onto slides. Slides were washed in 5% BSA and then incubated with primary rat anti-mouse F4/80 (Clone BM8; eBioscience) overnight at 4°C and then stained by the corresponding secondary antibody for 2 h. Cell nuclei were stained with 5 μg/mL 4,6-diamidino-2-phenylindole (DAPI). Images were acquired using an Olympus fluorescence microscope.
Hemin-platelets binding kinetic characterization
The binding kinetics of hemin to human platelets were measured using the bio-layer interferometry (BLI) biosensor (Gator Bio, USA). Briefly, hemin (100 nM) was loaded onto the sensors (SA, Gator Bio) and exposed to 62.5–1000 nM Recombinant Human CD42b/GP1BA Protein (His Tag) (HPLC-verified, Sino Biological, Cat: 11765-H08H) for association and dissociation with PBST buffer. The binding affinity was measured by the wavelength shift (nm) and used as the binding ratio to compare the binding affinities between the different groups. The binding ability was normalized to that of the relative control under the same conditions.
Statistical analysis
Values were represented as the mean ± standard deviation (SD). Statistical comparisons between different treatments were shown in the respective figure legends. Differences with p values less than 0.05 were considered significant.
Results
Extracellular hemin activates platelets
To investigate the platelet activity under hemolytic diseases, the effects of extracellular hemin on human platelets were studied in vitro. Our findings showed that hemin raised the expression of P-selectin upon platelet activation in a dose-dependent manner ( and S1). PS exposure was evaluated by measuring annexin V binding. Hemin significantly increased annexin V binding, however, by less than 5%, even at the highest concentration ( and S2), demonstrating PS exposure or generation of a procoagulant surface. Simultaneously, increased platelet aggregation at high concentrations was observed using light transmission aggregometry (). Moreover, platelet aggregometry was directly observed using confocal microscopy (). Larger platelet aggregates were observed in high-concentration hemin-treated platelets.
Figure 1. Hemin induced human platelet activation. (A, B) P-selectin and annexin V binding expression on the surface of hemin-treated platelets analyzed by FACS. The positive gate and specificity of these antibodies were controlled by corresponding isotypes. (C) Percentage aggregation measured using light transmission aggregometry in response to TRAP-6 (1.5 μM). Data are presented as the mean ± SD (panels A, B, and C); n = 3. *Compared with the hemin-free group (0 μM) (p<.05). Statistical analysis by one-way ANOVA with Tukey’s multiple comparison in panels A, B, and C. (D) Fluorescent images showing the formation and size of aggregates after incubation with hemin for 1 h. (E) Ten representative traces of intracellular Ca2+ dynamics in platelets treated with hemin. (F) Intracellular [Ca2+] transient amplitudes quantified by measuring the maximal fluorescence intensity change of fluo-4 AM in control and hemin-treated platelets. Data are presented as the mean ± SD (panels F); n = 10. *Compared with the hemin-free group (0 μM) (p<.05). Statistical analysis by independent-sample t-tests in panel F. (G) SEM observation of hemin-treated platelets in vitro. Scale bars = 10 μm. (H) TEM observation for studying platelet structure. Hemin (0 μM) represented resting human platelets; hemin (5–50 μM) represented hemin-activated human platelets. Scale bars = 2 μm. The pictures shown in G and H are representative of all experiments conducted. (I) Cytoskeleton staining in platelets. Washed platelets were treated with hemin and analyzed by confocal microscopy. Representative actin staining with phalloidin (red) and tubulin stained with anti-β-tubulin antibodies (green) in platelets preincubated with 0, 5, 25, and 50 μM hemin, respectively (scale bars = 20 μm).
![Figure 1. Hemin induced human platelet activation. (A, B) P-selectin and annexin V binding expression on the surface of hemin-treated platelets analyzed by FACS. The positive gate and specificity of these antibodies were controlled by corresponding isotypes. (C) Percentage aggregation measured using light transmission aggregometry in response to TRAP-6 (1.5 μM). Data are presented as the mean ± SD (panels A, B, and C); n = 3. *Compared with the hemin-free group (0 μM) (p<.05). Statistical analysis by one-way ANOVA with Tukey’s multiple comparison in panels A, B, and C. (D) Fluorescent images showing the formation and size of aggregates after incubation with hemin for 1 h. (E) Ten representative traces of intracellular Ca2+ dynamics in platelets treated with hemin. (F) Intracellular [Ca2+] transient amplitudes quantified by measuring the maximal fluorescence intensity change of fluo-4 AM in control and hemin-treated platelets. Data are presented as the mean ± SD (panels F); n = 10. *Compared with the hemin-free group (0 μM) (p<.05). Statistical analysis by independent-sample t-tests in panel F. (G) SEM observation of hemin-treated platelets in vitro. Scale bars = 10 μm. (H) TEM observation for studying platelet structure. Hemin (0 μM) represented resting human platelets; hemin (5–50 μM) represented hemin-activated human platelets. Scale bars = 2 μm. The pictures shown in G and H are representative of all experiments conducted. (I) Cytoskeleton staining in platelets. Washed platelets were treated with hemin and analyzed by confocal microscopy. Representative actin staining with phalloidin (red) and tubulin stained with anti-β-tubulin antibodies (green) in platelets preincubated with 0, 5, 25, and 50 μM hemin, respectively (scale bars = 20 μm).](/cms/asset/ef7743c4-7ca8-4e71-9a1a-260dbf7e12e7/iplt_a_2383642_f0001_oc.jpg)
Hemin can induce apoptosis and necroptosis in RBCs by increasing intracellular Ca2+ levels and eliminating cellular glutathione.Citation23 High intracellular Ca2+ enhances dynein activity and suppresses kinesin activity, producing a dynamic and rapid change in the cytoskeleton. In our study, we observed that a rapid Ca2+ burst occurred upon the addition of 25 μM hemin (). Changes in the platelet ultrastructure were also observed using SEM () and TEM (). As shown in . platelet morphology changed and pseudopodia extended after hemin treatment, reflecting the increased degree of activation, which was consistent with the increased expression of CD62P in . First, typical and distinguishable platelets with a discoid shape were predominant in the hemin-free group. Subsequently, 5 μM hemin stimulation facilitated cell adhesion. Thereafter, filopodia in 25 μM hemin-treated platelets formed a spindle-like morphology. However, in the case of 50 μM, lamellipodia filled the area between individual filopodia before a so-called “fried-egg” morphology was formed.
TEM analysis revealed that untreated platelets had a discoid shape, and granules were heterogeneously distributed in the cytoplasm (). Furthermore, 5 μM hemin-treated platelets displayed minor shape changes, retaining the basic discoid form to some extent. Most of the cells formed short pseudopods with evident centralization of α-granules, and the integrity of granules was moderately maintained and visible throughout the cytoplasm. Upon 25 μM hemin stimulation, the classic activation signs emerged and dramatic alterations in shape and noticeable degranulation, such as loss of the discoid shape, longer pseudopodia formation, enlargement of vacuoles, internal contraction of the platelet marginal band and aggregate formation with reduced cytoplasmic granules, were observed. In the 50 μM group, large vacuoles were seen in nearly all the platelets.
We analyzed the cytoskeleton dynamics in hemin-treated platelets using confocal microscopy. As shown in , resting platelets maintained their discoid shape with a well-demarcated and highly specialized cytoskeleton, while hemin treatment caused cytoskeleton reorganization, inducing depolymerization of the microtubule coil and loss of the discoid shape of platelets compared to controls. Further analysis suggested that hemin-treated platelets contracted and elongated, forming stress fibers, filopodia, and lamellipodia. Altogether, hemin-activated platelets increased intracellular Ca2+ and induced platelet PS exposure or the generation of a procoagulant surface apoptosis and morphological changes (). However, from a macro perspective, the morphological changes of platelets were hemin concentration-dependent and gradual.
Hemin-inhibited platelet clearance in vivo
In general, the clearance of apoptotic cells by macrophages plays a central role in tissue homeostasis. BMDMs are typically used to investigate the phagocytosis of apoptotic cells. Because macrophages are critical for clearing aged platelets,Citation16 we used BMDMs for phagocytosis experiments to simulate the platelet clearance process in vivo. FACS analysis revealed that after 4 h of co-incubation, hemin inhibited GFP+ mouse platelet phagocytosis by tdTomato BMDMs compared to the hemin-free group (). This observation was also confirmed by analyzing GFP+ platelets treated with hemin (0–50 μM) localization after 4 h of co-culture of tdTomato BMDMs by confocal microscopy. shows colocalization images of GFP+ platelets (green) and tdTomato BMDMs (red). The effect of hemin on the lifespan of platelets in vivo was investigated, and the post-transfusion recovery of hemin-treated platelets was evaluated by transfusing GFP+ mouse platelets into wild-type C57BL/6J mice. The percentage of GFP+ platelets from among the total platelets 20 min after transfusion was considered as 100% recovery in circulation. Notably, after 25 or 50 μM hemin-treatment, the platelets were cleared slowly at 24 h and 48 h, which was consistent with the results in vitro (). Furthermore, routine blood tests of blood samples from mice transfused with hemin-treated platelets showed no significant differences in platelet count compared to the control group at different timepoints (), suggesting that treatment with hemin affected the fate of platelets in vivo.
Figure 2. Treatment with hemin inhibited the clearance of mouse platelets in vivo. (A, B, C) phagocytosis of GFP+ platelets (green) by BMDMs (red) quantified by FACS (A, B) and confocal microscopy (C) in vitro. (D) Percentage clearance of GFP+ platelets after intravenous injection of GFP+ hemin-opsonized platelets. (E) Routine blood test results of platelets (PLT) number in different groups. (F, G) tissue sections of the liver (F) and spleen (G) harvested from mice stained with anti-F4/80 (liver and spleen macrophages; red). Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI) (blue). F: white scale bars, 50 μm. G: white scale bars, 40 μm. Data are presented as the mean ± SD; n = 3–5. *Compared with the hemin-free group (0 μM) (p<.05). Statistical analysis by one-way ANOVA with Tukey’s multiple comparison in panel A. Statistical analysis by two-way ANOVA with Tukey’s multiple comparison in panels D and E.
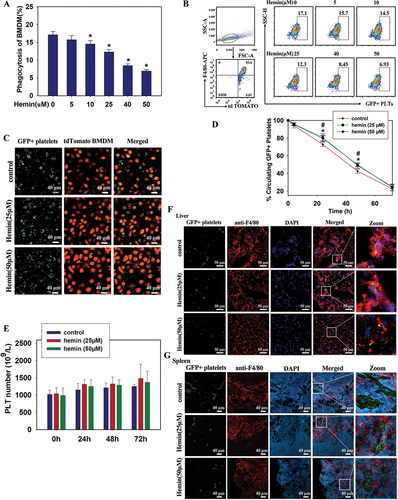
Next, tissues from recipient mice were examined after intravenous injection of GFP+ platelets by immunofluorescence staining. As expected, GFP+ platelets appeared in the liver and spleen (). Labeling macrophages with monoclonal antibody F4/80 revealed that most platelets were colocalized with macrophages in the liver, while a few were colocalized with macrophages in the spleen. These results suggest that macrophages play a key role in the clearance of hemin-treated platelets.
Hemin binds to GPIbα
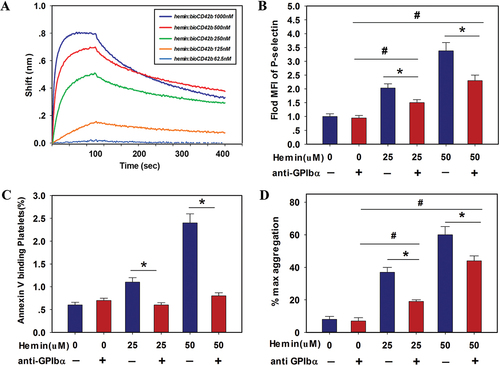
The molecular mechanisms underlying hemin-induced platelet activation were examined to elucidate the role of hemin in platelet clearance. Hemoglobin induced platelet activation and apoptosis through interactions with surface GPIbα,Citation4 suggesting GPIbα as the main receptor of hemin. The BLI biosensor was used to determine the binding affinity between hemin and GPIbα. Hemin bound to GPIbα with high affinities; the affinity constant (KD) of GPIbα to hemin was 15.7 nM (). We then investigated the role of GPIbα signaling in hemin-treated human platelet activation. The pretreatment of platelets with HIP1, an anti-GPIbα antibody, to block GPIbα prevented platelet aggregation,Citation24 inhibiting hemin-induced activation, apoptosis, and aggregation of platelets to some degree (). Collectively, these results allowed us to speculated that hemin could probably bind to GPIbα on the platelet surface and it might partly activate platelets this way. Simultaneously, it is worth noting that the downstream signalings such as P-selectin expression and PS exposure, were not inhibited completely. Thus, we suspect that there was other pathways involved in the binding events in hemin treating.
Figure 3. Hemin induced human platelet activation by binding to GPIbα. (A) The binding kinetic characteristics of the hemin-platelet complex were detected by the BLI biosensor. (B-D) blocking effect of anti-GPIbα antibody (H1P1) (20 μg/mL) on hemin (0, 25, 50 μM)-induced platelet activation, PS exposure and aggregation. Washed platelets were pretreated with anti-GPIbα antibody and incubated with hemin. The expressions of P-selectin (B), and annexin V binding (C) on the platelet surface were measured by FACS. The positive gate and specificity of these antibodies were controlled by corresponding isotypes. (D) Quantification of maximum light transmission of the platelets. Data are presented as the mean ± SD; n = 3. *Compared with the anti-GPIbα antibody-free group (p<.05). #Compared with anti-GPIbα antibody (5 μg/mL)/hemin-free group (p<.05). Statistical analysis by two-way ANOVA with Tukey’s multiple comparison in panel B-E.
Hemin-mediated reduction in human platelet desialylation attenuated platelet clearance
Previous studies have demonstrated that GPIbα plays a pivotal role in the rapid clearance of transfused cold-stored platelets.Citation18 Therefore, we evaluated the expression of GPIbα in hemin-treated platelets. Hemin treatment could down-regulate the surface expression of GPIbα (CD42b). Notably, FACS analysis revealed that as hemin concentrations increased, GPIbα surface expression decreased (). However, Western blot data showed no significant difference in platelet GPIbα levels after hemin treatment (), indicating that total GPIbα remained unchanged.
Figure 4. Hemin inhibited human platelet clearance via decreased desialylation of GPIbα (A, B) the expression level of GPIbα in hemin-treated human platelets assessed by FACS using FITC-conjugated CD42b. (C) Western blot analysis of GPIbα. (D) FACS analysis of mitochondrial membrane potential (ΔΨm) in human platelets. (E) FACS analysis of ROS generation. (F) Hemin inhibited GPIbα desialylation of human platelets. The positive gate and specificity of these antibodies were controlled by corresponding isotypes. Data are presented as the mean ± SD; n = 3. *Compared with the hemin-free group (0 μM) (p<.05). Statistical analysis by one-way ANOVA with Tukey’s multiple comparison in panels A, D, E, and F. (G) Blocking GPIbα inhibited platelet clearance. GFP+ mouse platelets pre-incubated with anti-GPIbα blocking antibody (20 μg/mL) for 30 min prior to hemin treatment, then phagocytosis of GFP+ platelets by BMDMs was quantified by FACS. Data are presented as the mean ± SD; n = 3. *Compared with the anti-GPIbα antibody-free group (p<.05), #Compared with the anti-GPIbα antibody (5 μg/mL)/hemin-free group (p<.05). Statistical analysis by two-way ANOVA with Tukey’s multiple comparison in panel G.
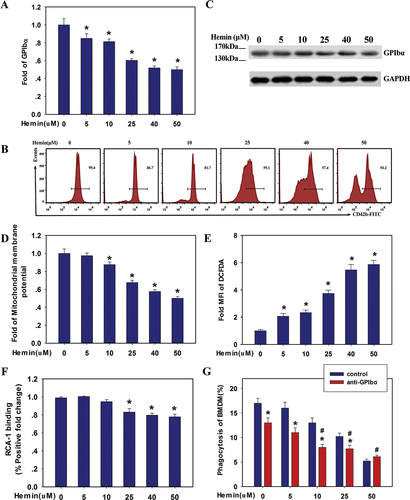
Previous studies have shown that mitochondrial ROS play a critical role in DOX-induced platelet apoptosis and GPIbα shedding,Citation25 and that ROS play a crucial role in thrombin and collagen-induced GPIbα ectodomain shedding.Citation26 Therefore, we analyzed mitochondrial membrane activity in the presence of different concentrations of hemin (5–50 μM). As shown in , human platelets treated with hemin showed reduced mitochondrial membrane potential and increased ROS generation in a dose-dependent manner.
Because hemin bound to GPIbα with high affinity, we tested the effect of hemin treatment on platelet GPIbα desialylation by quantifying the binding of fluorescein-conjugated RCA-1 lectins, which specifically target exposed galactose residues following desialylation.Citation17,Citation18 RCA-1 binding to human platelets was reduced and desialylation significantly reduced after hemin treatment ().
To clarify the role of platelet GPIbα expression in hemin-treated platelet clearance by macrophages, we then examined the mouse platelet clearance by BMDMs after anti-GPIbα antibody pretreatment of platelets in vitro. Phagocytosis of platelets by BMDMs was reduced in low concentration hemin groups (0, 5 and 10 μM) to a certain extent after GPIbα blocking. However, it was worth noting that the role of GPIb blocking disappeared in the case of 50 μM hemin group (), indicating that GPIbα is involved in platelet clearance. It is reasonable to deduce that the dose-dependent activation and aggregation of platelets toward hemin exposure probably accelerate the clearance of platelets by BMDM, neutralizing the effect of GPIba blocking, especially in the high levels hemin groups. We speculate that besides the GPIbα desialylation pathway, other clearance mechanisms might also be at work in hemin-mediated platelet clearance.
Hemin may inhibit the desialylation of GPIbα through binding or occupation, which might affect its simultaneous recognition by macrophages and hepatocytes and in turn slow down the platelet clearance in vivo ().
Discussion
Previous studies have shown that hemin is a ligand of various platelet receptors such as CLEC-2, GPVI,Citation8 and TLR4, and activates human and mouse platelets. In this study, we analyzed the effect of hemin on platelets platelet activation and clearance through in vitro and in vivo experiments. BLI analysis and KD value of 15.7 nM after platelet treatment suggested high affinity binding between hemin and GPIbα on platelets. Moreover, anti-GPIbα antibody pretreatment inhibited hemin-induced platelet activation, indicating that hemin binds GPIbα, reinforcing that GPIbα mainly could mediate the effect of hemin on platelets. Previous studies have shown that VWF-GPIb-IX interaction induces platelet activation, leading to platelet adhesion and aggregation.Citation27,Citation28 Similarly, in this study, we found that hemin (5–50 μM) induced platelet activation in a concentration-dependent manner in vitro. Hemin can induce apoptosis and necroptosis in RBCs by increasing intracellular Ca2+ levels and eliminating cellular glutathione.Citation23 High intracellular Ca2+ enhances dynein activity while reducing kinesin activity, producing a dynamic and rapid change in the cytoskeleton. Another study showed that myeloperoxidase binding to platelets induced actin cytoskeleton reorganization that elevated cytosolic Ca2+ concentrations.Citation29 Therefore, hemin-GPIbα interaction was speculated to initiate cytoskeleton reorganization to increase [Ca2+]i. As expected, in the current study high [Ca2+]i enhanced microtubule coil expansion and produced dynamic and rapid changes in the cytoskeleton. Our data also showed that hemin-activating platelets induced changes in platelet shape, including the formation of filopodia and lamellipodia from actin filaments and the dispersal of microtubule ring fragments, indicating that a hemin-dependent increase in [Ca2+]i likely regulates redistribution of the cytoskeleton network in platelets, leading to the formation of larger aggregates, which marks a procoagulant state and thrombosis.
Billions of platelets are cleared every day from circulation through efficient regulatory mechanisms, which can be regulated by a variety of factors, such as exogenous reagents or environmental changes. Two case reports have shown that the platelet count decreases after hemin injection.Citation30,Citation31 In contrast, a study in rats showed that platelet count increased after an intraperitoneal injection of hemin.Citation32 In our study, we found that platelet count decreased after hemin treatment in vitro (Fig. S3), probably due to increased platelet aggregation. However, when residual hemin-treated platelets were injected intravenously into mice, the post-transfusion recovery of these platelets rose in vivo at 24 h and 48 h after transfusion, indicating that the hemin-treated platelets cleared slowly. The in vitro data also showed that the presence of hemin inhibited platelet phagocytosis by BMDMs. However, this is inconsistent with the findings of previous studies that show platelet clearance from circulation is accelerated under (pro)thrombotic conditions once they have executed their functions.Citation33 We speculated that this bias might be because no significant aggregates or apoptosis were formed despite the apparent platelet activation observed in vitro.
Intersecting molecular mechanisms regulate platelet clearance. The binding of VWF or antibodies to the GPIbα on platelets can activate GPIb-IX, triggering downstream signaling in the platelet such as activation and PS exposure. It has been shown that the loss of terminal sialic acid (a derivative of neuraminic acid) from the platelet surface is directly correlated with senescent platelet clearance from circulation.Citation34 Deglycosylation of GPIbα, a subunit of GPIb, also causes hepatic clearance of cold-stored mice platelets after transfusion.Citation35,Citation36 We observed a significantly reduced surface GPIbα (CD42b) in platelets upon the addition of hemin, however, no significant changes in GPIbα expression were observed from western blot results. It is can be deduced that the reduced exposure of galactose residues may be due to the decreased sialic acid loss at the end of GPIba instead of GPIba extracellular domain loss, for there was no glycocalicin loss detected in the medium after hemin treatment (data not shown). Simultaneously, we speculated that hemin might have probably occupy the position of GPIbα instead of inducing the structural changes of it (such as cluster formation). It is known that glycan modification is involved in platelet clearance in human and mice after the stage of desialylation.Citation35,Citation37 In our study, a significantly reduced exposure of galactose residues (RCA-1) was detected, which indicated reduced GPIbα-mediated desialylation of hemin-treated platelets, however, we also can’t exclude the possibility that hemin remaining in the system after washing interfered with the binding of RCA-I to GPIbα. Notably, hemin treatment induced PS exposure, which was another mechanism that contributed to platelet clearance. However, despite the significant increase in platelet PS exposure after hemin treatment, in vitro and in vivo data showed that platelet clearance was slowed. Therefore, it was likely a balanced process and inhibition of desialylation might be dominating and responsible for the slowing of hemin-mediated platelet clearance.
Finally, previous studies have demonstrated that GPIbα clustering induces liver macrophage-mediated platelet clearance in guinea pigs and macaques.Citation24 And platelet clearance in immune thrombocytopenia (ITP) is mediated by FcγR via macrophages of the reticuloendothelial system through Fc - FcγR interactions primarily in the spleen.Citation38 We found that platelets were mainly cleared by macrophages in the liver, which is consistent with previous studies in rats and mice.Citation39,Citation40 In summary, hemin binding to GPIbα induced platelet activation, but inhibited platelet desialylation and in turn extended the circulation time of platelets, suggesting that the hemin-treated platelets by allogeneic transfusion obtained more recovery in our mouse model (). However, the connection between these processes remains unclear, which necessitate further experimental investigation.
Figure 5. Schematic representation of putative mechanisms underlying platelet activation and clearance in hemolytic disease induced by hemin. Hemin binding to GPIbα induced platelet activation, such as increased [Ca2+]i level, ROS generation, PS expression, and morphological changes, but inhibited platelet desialylation and slowed platelet clearance. Figure 5. Was created with BioRender.com.
![Figure 5. Schematic representation of putative mechanisms underlying platelet activation and clearance in hemolytic disease induced by hemin. Hemin binding to GPIbα induced platelet activation, such as increased [Ca2+]i level, ROS generation, PS expression, and morphological changes, but inhibited platelet desialylation and slowed platelet clearance. Figure 5. Was created with BioRender.com.](/cms/asset/a4f89d33-a21e-4b17-8247-4c6cc895fec1/iplt_a_2383642_f0005_oc.jpg)
Conclusion
In summary, our study shows that hemin plays an important role in regulating normal platelet clearance during severe hemolysis. Herein, we demonstrated that hemin activates platelets, as evidenced by downstream signaling pathways, such as: Increased intracellular Ca2+ levels, morphological changes, and platelet aggregation response in a dose-dependent manner. The binding of hemin to GPIbα on platelets could be involved in inducing platelet activation, which might slow platelet clearance by decreasing GPIbα desialylation in vivo. Further research is needed to elucidate the precise mechanism linking hemolysis to platelet activation and clearance regulation. Our findings highlight the need to focus on the status and function of platelets in the diagnosis and therapy of hemolytic disorders, which may, in turn, provide insights into novel clinical strategies.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- White NJ. Anaemia and malaria. Malar J. 2018;17(1):371. doi:10.1186/s12936-018-2509-9.
- Martins R, Maier J, Gorki AD, Huber KV, Sharif O, Starkl P, Saluzzo S, Quattrone F, Gawish R, Lakovits K, et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat Immunol. 2016;17(12):1361–11. doi:10.1038/ni.3590.
- Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, Smith A, Nath KA, Hebbel RP, Vercellotti GM. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. 2014;123(3):377–90. doi:10.1182/blood-2013-04-495887.
- Singhal R, Annarapu GK, Pandey A, Chawla S, Ojha A, Gupta A, Cruz MA, Seth T, Guchhait P. Hemoglobin interaction with GP1bα induces platelet activation and apoptosis: a novel mechanism associated with intravascular hemolysis. Haematologica. 2015;100(12):1526–33. doi:10.3324/haematol.2015.132183.
- Annarapu GK, Nolfi-Donegan D, Reynolds M, Wang Y, Kohut L, Zuckerbraun B, Shiva S. Heme stimulates platelet mitochondrial oxidant production to induce targeted granule secretion. Redox Biol. 2021;48:102205. doi:10.1016/j.redox.2021.102205.
- Zhang L. Heme biology: the secret life of heme in regulating diverse biological processes[J]. WORLD SCIENTIFIC; 2019.
- Shimizu T, Lengalova A, Martínek V, Martínková M. Heme: emergent roles of heme in signal transduction, functional regulation and as catalytic centres. Chem Soc Rev. 2019;48(24):5624–57. doi:10.1039/C9CS00268E.
- Oishi S, Tsukiji N, Otake S, Oishi N, Sasaki T, Shirai T, Yoshikawa Y, Takano K, Shinmori H, Inukai T, et al. Heme activates platelets and exacerbates rhabdomyolysis-induced acute kidney injury via CLEC-2 and GPVI/FcRγ. Blood Adv. 2021;5(7):2017–26. doi:10.1182/bloodadvances.2020001698.
- Burkhart JM, Vaudel M, Gambaryan S, Radau S, Walter U, Martens L, Geiger J, Sickmann A, Zahedi RP. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120(15):e73–82. doi:10.1182/blood-2012-04-416594.
- Du X, Beutler L, Ruan C, Castaldi PA, Berndt MC. Glycoprotein Ib and glycoprotein IX are fully complexed in the intact platelet membrane. Blood. 1987;69(5):1524–7. doi:10.1182/blood.V69.5.1524.1524.
- Lopez JA, Chung DW, Fujikawa K, Hagen FS, Davie EW, Roth GJ. The alpha and beta chains of human platelet glycoprotein Ib are both transmembrane proteins containing a leucine-rich amino acid sequence. Proc Natl Acad Sci USA. 1988;85(7):2135–9. doi:10.1073/pnas.85.7.2135.
- Savage B, Saldívar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84(2):289–97. doi:10.1016/S0092-8674(00)80983-6.
- Bryckaert M, Rosa JP, Denis CV, Lenting PJ. Of von Willebrand factor and platelets. Cell Mol Life Sci. 2015;72(2):307–26. doi:10.1007/s00018-014-1743-8.
- Okumura I, Lombart C, Jamieson GA. Platelet glycocalicin. II. Purification and characterization. J Biol Chem. 1976;251(19):5950–5. doi:10.1016/S0021-9258(17)33044-2.
- Grozovsky R, Begonja AJ, Liu K, Visner G, Hartwig JH, Falet H, Hoffmeister KM. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med. 2015;21(1):47–54. doi:10.1038/nm.3770.
- Deppermann C, Kratofil RM, Peiseler M, David BA, Zindel J, Castanheira F, van der Wal F, Carestia A, Jenne CN, Marth JD, et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J Exp Med. 2020;217(4). doi:10.1084/jem.20190723.
- Wandall HH, Hoffmeister KM, Sørensen AL, Rumjantseva V, Clausen H, Hartwig JH, Slichter SJ. Galactosylation does not prevent the rapid clearance of long-term, 4 degrees C-stored platelets. Blood. 2008;111(6):3249–56. doi:10.1182/blood-2007-06-097295.
- Hoffmeister KM, Josefsson EC, Isaac NA, Clausen H, Hartwig JH, Stossel TP. Glycosylation restores survival of chilled blood platelets. Science. 2003;301(5639):1531–4. doi:10.1126/science.1085322.
- Rauch A, Dupont A, Rosa M, Desvages M, Le Tanno C, Abdoul J, Didelot M, Ung A, Ruez R, Jeanpierre E, et al. Shear forces induced platelet clearance is a new mechanism of thrombocytopenia. Circ Res. 2023;133(10):826–41. doi:10.1161/CIRCRESAHA.123.322752.
- Quach ME, Chen W, Li R. Mechanisms of platelet clearance and translation to improve platelet storage. Blood. 2018;131(14):1512–21. doi:10.1182/blood-2017-08-743229.
- Muller-Eberhard U, Javid J, Liem HH, Hanstein A, Hanna M. Brief report: plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood. 1968;32(5):811–5. doi:10.1182/blood.V32.5.811.811.
- Gouveia Z, Carlos AR, Yuan X, Aires-da-Silva F, Stocker R, Maghzal GJ, Leal SS, Gomes CM, Todorovic S, Iranzo O, et al. Characterization of plasma labile heme in hemolytic conditions. FEBS J. 2017;284(19):3278–301. doi:10.1111/febs.14192.
- Gladwin MT. Cardiovascular complications and risk of death in sickle-cell disease. The Lancet. 2016;387(10037):2565–74. doi:10.1016/S0140-6736(16)00647-4.
- Yan R, Chen M, Ma N, Zhao L, Cao L, Zhang Y, Zhang J, Yu Z, Wang Z, Xia L, et al. Glycoprotein Ibα clustering induces macrophage-mediated platelet clearance in the liver. Thromb Haemost. 2015;113(1):107–17. doi:10.1160/TH14-03-0217.
- Wang Z, Wang J, Xie R, Liu R, Lu Y. Mitochondria-derived reactive oxygen species play an important role in doxorubicin-induced platelet apoptosis. Int J Mol Sci. 2015;16(5):11087–100. doi:10.3390/ijms160511087.
- Zhang P, Du J, Zhao L, Wang X, Zhang Y, Yan R, Dai J, Liu G, Zhang F, Dai K. The role of intraplatelet reactive oxygen species in the regulation of platelet glycoprotein Ibα ectodomain shedding. Thromb Res. 2013;132(6):696–701. doi:10.1016/j.thromres.2013.09.034.
- Kim J, Zhang CZ, Zhang X, Springer TA. A mechanically stabilized receptor-ligand flex-bond important in the vasculature. Nature. 2010;466(7309):992–5. doi:10.1038/nature09295.
- Liu J, Fitzgerald ME, Berndt MC, Jackson CW, Gartner TK. Bruton tyrosine kinase is essential for botrocetin/VWF-induced signaling and GPIb-dependent thrombus formation in vivo. Blood. 2006;108(8):2596–603. doi:10.1182/blood-2006-01-011817.
- Gorudko IV, Sokolov AV, Shamova EV, Grudinina NA, Drozd ES, Shishlo LM, Grigorieva DV, Bushuk SB, Bushuk BA, Chizhik SA, et al. Myeloperoxidase modulates human platelet aggregation via actin cytoskeleton reorganization and store-operated calcium entry. Biol Open. 2013;2(9):916–23. doi:10.1242/bio.20135314.
- Gajra A, Vajpayee N, Singh M, Coyle TE, Wright J. Hematin induced coagulopathy in acute intermittent porphyria: a case report. Blood. 2000;96(11 PART II):82b.
- Glueck R, Green D, Cohen I, Ts’ao CH. Hematin: unique effects of hemostasis. Blood. 1983;61(2):243–9. doi:10.1182/blood.V61.2.243.243.
- Desbuards N, Rochefort GY, Schlecht D, Machet M, Halimi J, Eder V, Hyvelin J, Antier D. Heme oxygenase-1 inducer hemin prevents vascular thrombosis. Thromb Haemost. 2007;98(3):614–20.
- Kim OV, Nevzorova TA, Mordakhanova ER, Ponomareva AA, Andrianova IA, Le Minh G, Daminova AG, Peshkova AD, Alber MS, Vagin O, et al. Fatal dysfunction and disintegration of thrombin-stimulated platelets. Haematologica. 2019;104(9):1866–78. doi:10.3324/haematol.2018.202309.
- Soslau G, Giles J. The loss of sialic acid and its prevention in stored human platelets. Thromb Res. 1982;26(6):443–55. doi:10.1016/0049-3848(82)90316-4.
- Rumjantseva V, Grewal PK, Wandall HH, Josefsson EC, Sørensen AL, Larson G, Marth JD, Hartwig JH, Hoffmeister KM. Dual roles for hepatic lectin receptors in the clearance of chilled platelets. Nat Med. 2009;15(11):1273–80. doi:10.1038/nm.2030.
- Sørensen AL, Rumjantseva V, Nayeb-Hashemi S, Clausen H, Hartwig JH, Wandall HH, Hoffmeister KM. Role of sialic acid for platelet life span: exposure of beta-galactose results in the rapid clearance of platelets from the circulation by asialoglycoprotein receptor-expressing liver macrophages and hepatocytes. Blood. 2009;114(8):1645–54. doi:10.1182/blood-2009-01-199414.
- Li Y, Fu J, Ling Y, Yago T, McDaniel JM, Song J, Bai X, Kondo Y, Qin Y, Hoover C, et al. Sialylation on O-glycans protects platelets from clearance by liver Kupffer cells. Proc Natl Acad Sci USA. 2017;114(31):8360–5. doi:10.1073/pnas.1707662114.
- McMillan R. The pathogenesis of chronic immune thrombocytopenic purpura. Semin Hematol. 2007;44(4 Suppl 5):S3–s11. doi:10.1053/j.seminhematol.2007.11.002.
- Casari C, Du V, Wu YP, Kauskot A, de Groot PG, Christophe OD, Denis CV, de Laat B, Lenting PJ. Accelerated uptake of VWF/platelet complexes in macrophages contributes to VWD type 2B-associated thrombocytopenia. Blood. 2013;122(16):2893–902. doi:10.1182/blood-2013-03-493312.
- Sanders WE, Read MS, Reddick RL, Garris JB, Brinkhous KM. Thrombotic thrombocytopenia with von Willebrand factor deficiency induced by botrocetin. An animal model. Lab Invest. 1988;59:443–52.